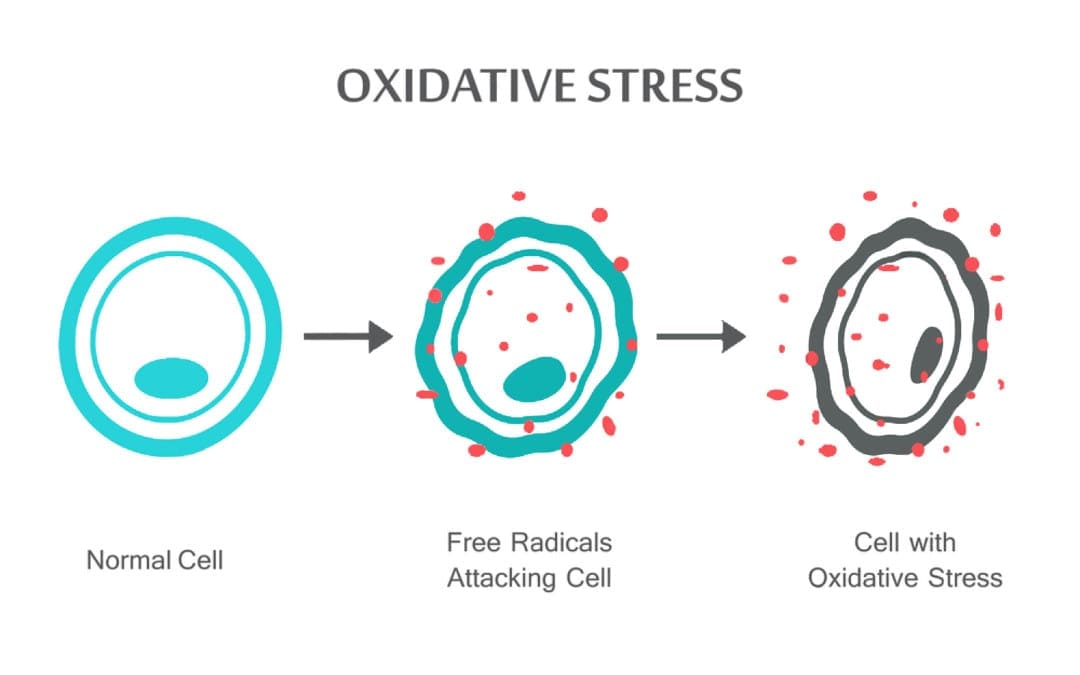
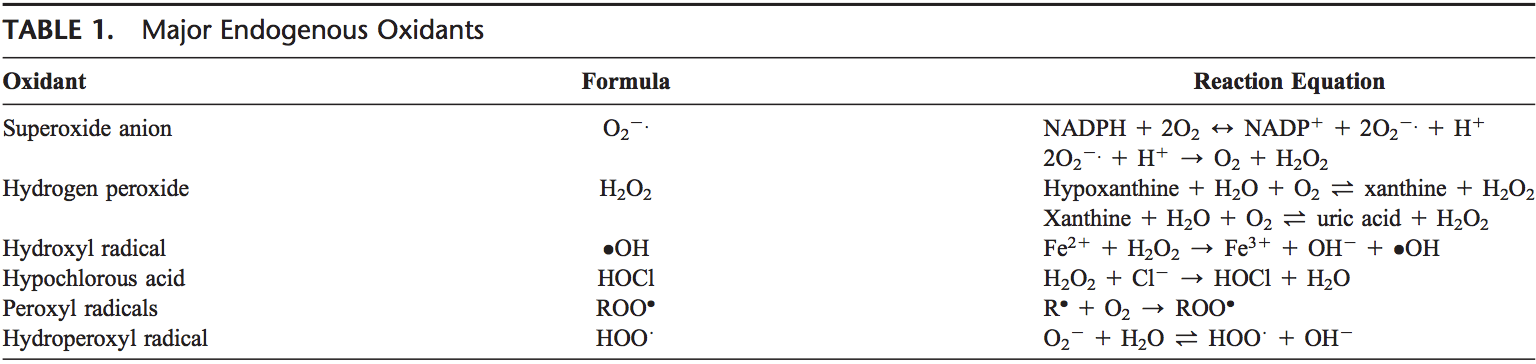
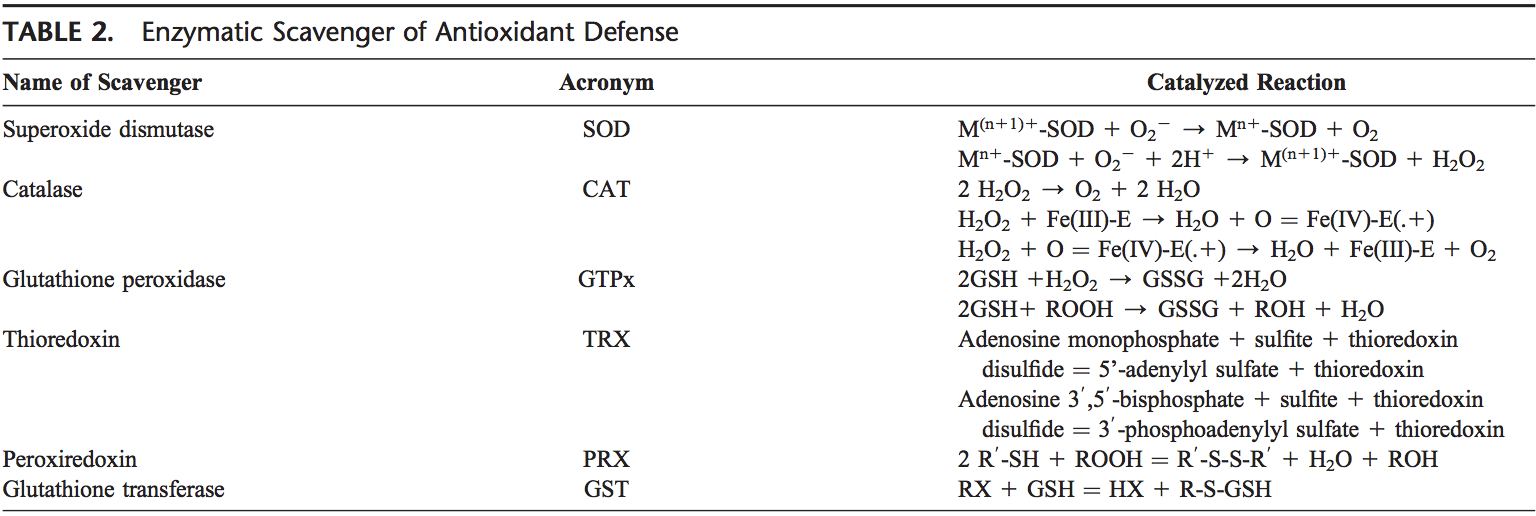
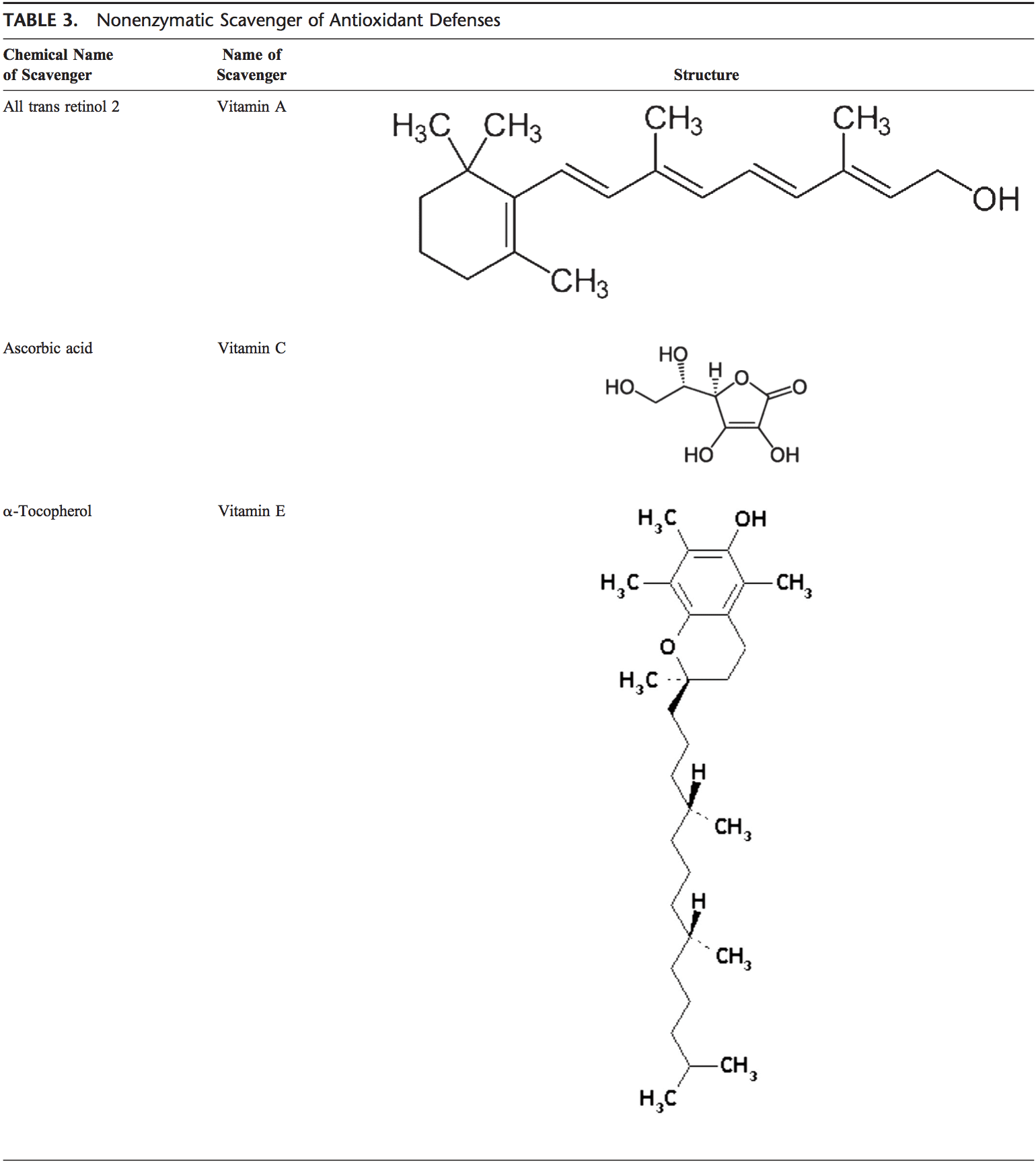
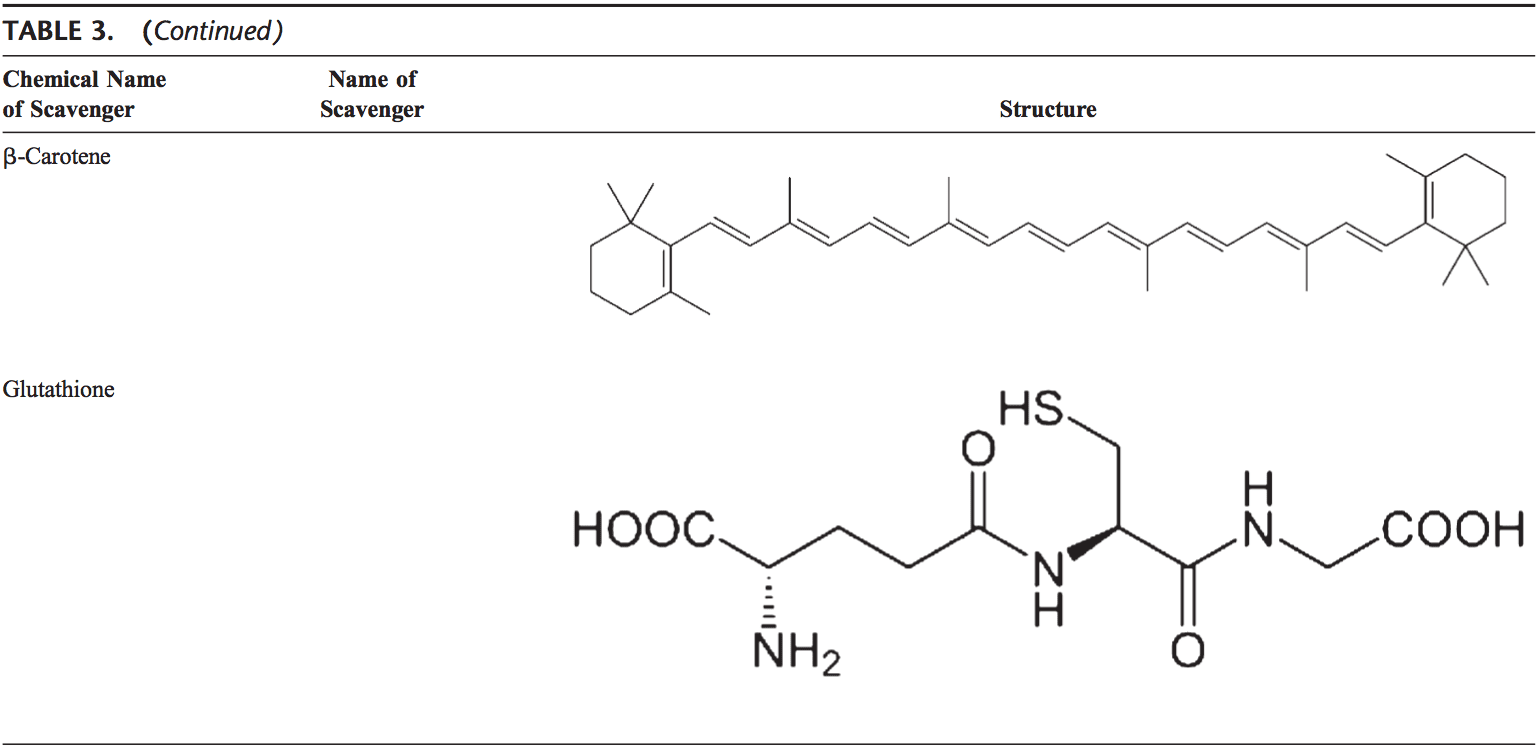
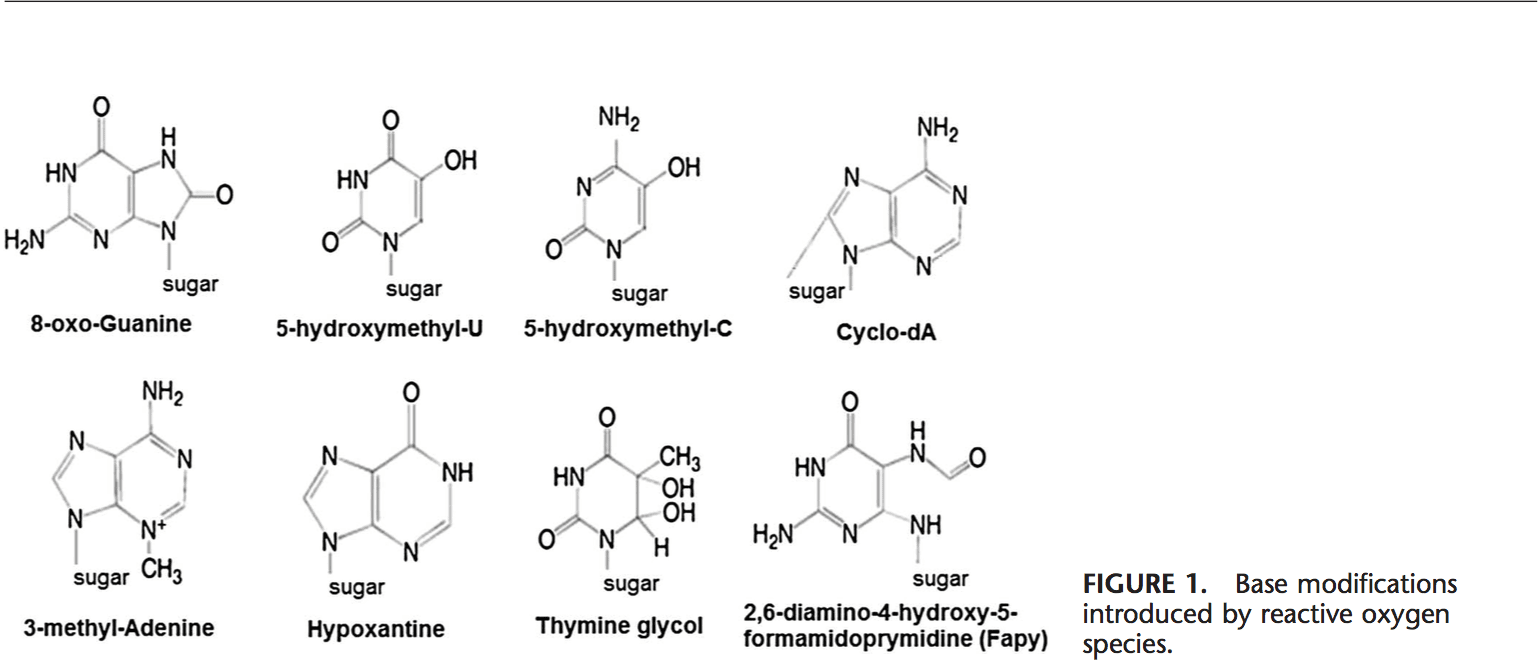
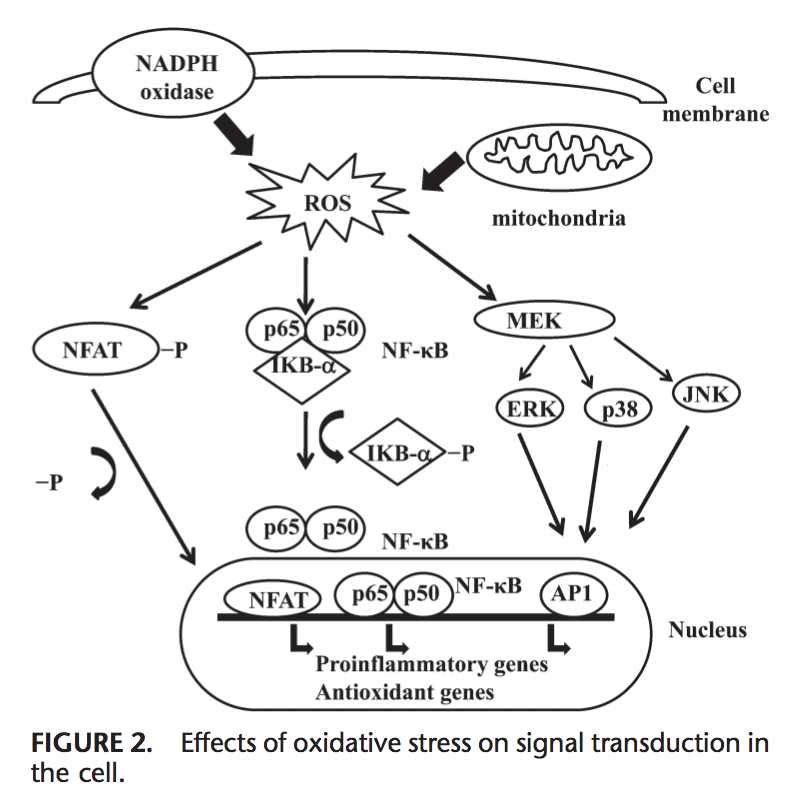
Equipo de Quiropráctica e Medicina Funcional de Estrés Oxidativo da Clínica Atrás. O estrés oxidativo defínese como unha perturbación no equilibrio entre a produción de osíxeno reactivo (radicais libres) e as defensas antioxidantes. Noutras palabras, é un desequilibrio entre a produción de radicais libres e a capacidade do organismo para contrarrestar ou desintoxicar os efectos nocivos mediante a neutralización mediante antioxidantes. O estrés oxidativo leva a moitas condicións fisiopatolóxicas no corpo. Estes inclúen enfermidades neurodexenerativas, é dicir, enfermidade de Parkinson, enfermidade de Alzheimer, mutacións xenéticas, cancro, síndrome de fatiga crónica, síndrome do X fráxil, trastornos do corazón e dos vasos sanguíneos, aterosclerose, insuficiencia cardíaca, ataque cardíaco e enfermidades inflamatorias. A oxidación ocorre baixo unha serie de circunstancias:
as células usan a glicosa para facer enerxía
o sistema inmunitario combate as bacterias e crea inflamación
os organismos desintoxican os contaminantes, os pesticidas e o fume de cigarro
Hai millóns de procesos que se producen nos nosos corpos en calquera momento que poden producir oxidación. Aquí tes algúns síntomas:
Fatiga
Falta de memoria e néboa cerebral
Dolores musculares ou articulares
Engurras xunto co pelo gris
Diminución da vista
Dores de cabeza e sensibilidade ao ruído
Susceptibilidade ás infeccións
Elixir alimentos orgánicos e evitar as toxinas no teu ambiente fai unha gran diferenza. Isto, xunto coa redución do estrés, pode ser beneficioso para diminuír a oxidación.
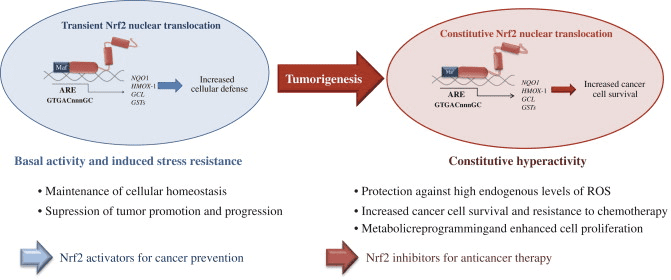
Os oxidantes generalmente prodúcense de forma controlada para regular procesos esenciais no corpo humano, incluíndo a división celular, a inflamación, a función inmune, a autofagia ea resposta ao estrés. Non obstante, a produción incontrolada destes oxidantes pode contribuír estrés oxidativo, que pode afectar a función celular, o que conduce ao desenvolvemento da toxicidade, enfermidades crónicas e cancro. Os mecanismos antioxidantes protectores do corpo humano están regulados por unha serie de vías vitais que controlan a resposta da célula aos oxidantes. O factor nuclear eritroide 2 factor, tamén coñecido como Nrf2, é un regulador emerxente de resistencia móbil aos oxidantes. O obxectivo do artigo seguinte é discutir e demostrar o papel emerxente de Nrf2 na función mitocondrial.
Abstracto
O factor de transcrición NF-E2 factor 45 relacionado con p2 (Nrf2; nome xenético NFE2L2) permite a adaptación e supervivencia en condicións de estrés regulando a expresión xénica de diversas redes de proteínas citoprotectoras, incluíndo encimas antioxidantes, antiinflamatorias e de desintoxicación. como proteínas que axudan na reparación ou eliminación de macromoléculas danadas. Nrf2 ten un papel crucial no mantemento da homeostase redox celular ao regular a biosíntese, a utilización e a rexeneración de glutatión, tiorredoxina e NADPH e controlando a produción de especies reactivas de osíxeno polas mitocondrias e a NADPH oxidase. En condicións homeostáticas, o Nrf2 afecta o potencial da membrana mitocondrial, a oxidación dos ácidos graxos, a dispoñibilidade de substratos (NADH e FADH2/succinato) para a respiración e a síntese de ATP. En condicións de estrés ou estimulación do factor de crecemento, a activación de Nrf2 contrarresta o aumento da produción de especies reactivas de osíxeno nas mitocondrias mediante a regulación transcripcional da proteína de desacoplamento 3 e inflúe na bioxénese mitocondrial mantendo os niveis de factor respiratorio nuclear 1 e receptor activado polo proliferador de peroxisomas. coactivador 1?, así como promovendo a biosíntese de nucleótidos de purina. Os activadores farmacolóxicos Nrf2, como o isotiocianato sulforafano natural, inhiben a apertura mediada por oxidantes do poro de transición da permeabilidade mitocondrial e o inchazo mitocondrial. Curiosamente, descubriuse que un composto sintético de 1,4-difenil-1,2,3-triazol, deseñado orixinalmente como activador de Nrf2, promove a mitofaxia, contribuíndo así á homeostase mitocondrial global. Así, Nrf2 é un actor destacado no apoio á integridade estrutural e funcional das mitocondrias, e este papel é particularmente crucial en condicións de estrés.
Nrf2 ten un papel crucial no mantemento da homeostase redox móbil.
Nrf2 afecta o potencial da membrana mitocondrial ea síntese de ATP.
Nrf2 inflúe na oxidación mitocondrial de ácidos graxos.
Nrf2 soporta a integridade estrutural e funcional das mitocondrias.
Os activadores de Nrf2 teñen efectos beneficiosos cando a función mitocondrial está comprometida.
introdución
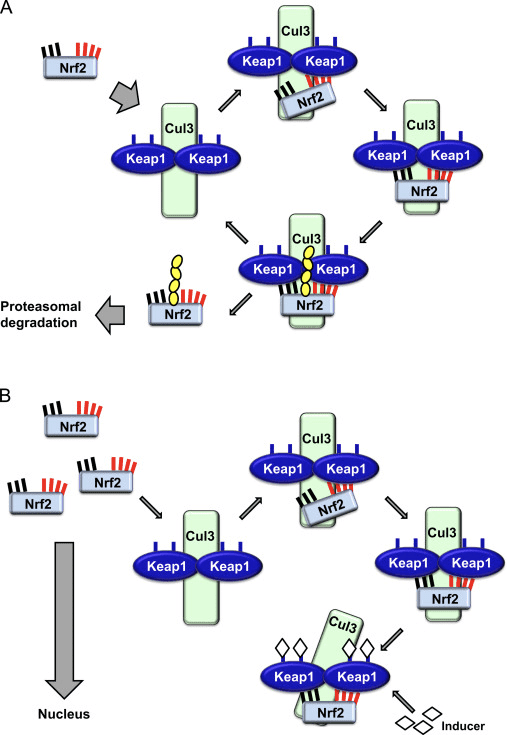
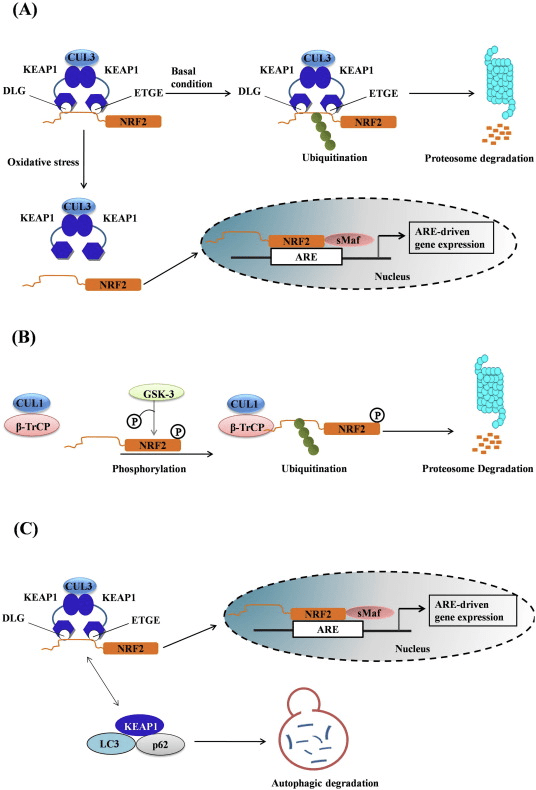
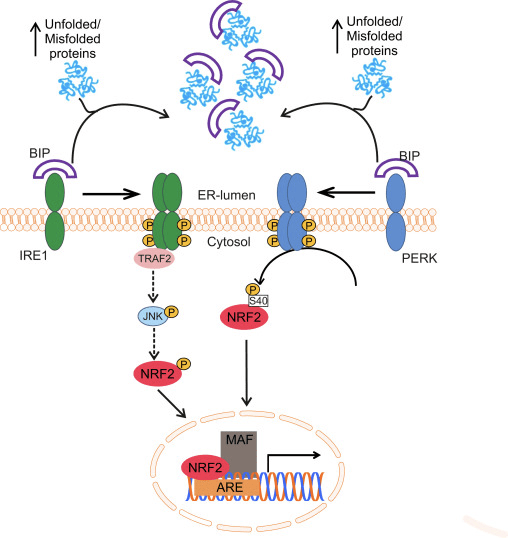
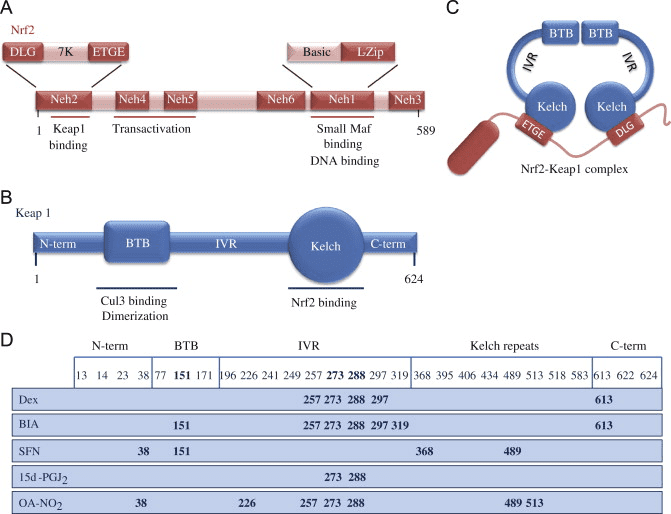
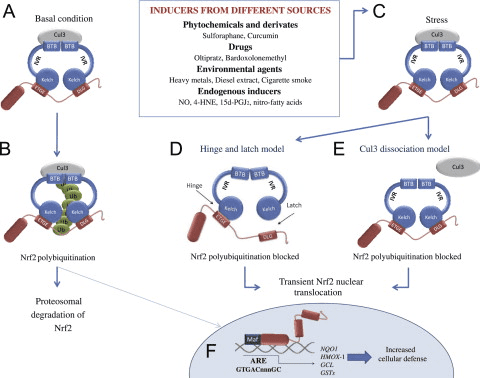
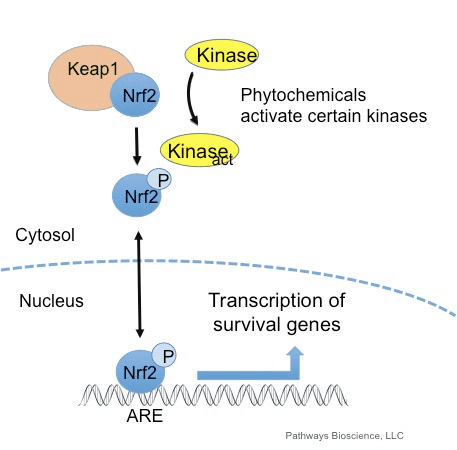
O factor de transcrición NF-E2 factor 45 relacionado con p2 (Nrf2; nome xenético NFE2L2) regula a expresión de redes de xenes que codifican proteínas con diversas actividades citoprotectoras. O propio Nrf2 está controlado principalmente a nivel de estabilidade da proteína. En condicións basais, Nrf2 é unha proteína de curta duración que está sometida a unha continua ubiquitinación e degradación proteasómica. Hai tres sistemas de ubiquitina ligase coñecidos que contribúen á degradación de Nrf2. Históricamente, o primeiro regulador negativo de Nrf2 que se descubriu foi a proteína 1 asociada a ECH tipo Kelch (Keap1) [1], unha proteína adaptadora de substrato para Cullin 3 (Cul3)/Rbx1 ubiquitin ligase [2], [3], [4], [1], [2] 1]. Keap1 utiliza un mecanismo cíclico altamente eficiente para dirixirse a Nrf5 para a ubiquitinación e a degradación do proteasoma, durante o cal Keap2 rexenerase continuamente, permitindo que o ciclo continúe (Fig. 3A) [1]. Nrf6 tamén está sometido a degradación mediada pola glicóxeno sintase quinase (GSK)7/?-TrCP dependente da ubiquitina ligase baseada en Cul2 [3], [1]. Máis recentemente, informouse de que, durante as condicións de tensión do retículo endoplasmático, Nrf8 está ubiquitinado e degradado nun proceso mediado pola ubiquitina ligase EXNUMX HrdXNUMX [XNUMX].
Figura 1 O modelo de unión e regeneración secuencial cíclico para a degradación mediada por Keap1 de Nrf2. (A) Nrf2 únese secuencialmente a un dímero Keap1 libre: primeiro a través do seu dominio de unión ETGE (pegada vermella) de alta afinidade e despois a través do seu dominio de unión DLG de baixa afinidade (varas negras). Nesta conformación do complexo proteico, Nrf2 sofre ubiquitinación e está dirixido a degradación proteasómica. O Keap1 libre é rexenerado e capaz de se ligar ao Nrf2 recén traducido eo ciclo comeza de novo. (B) Os indutores (diamantes brancos) reaccionan coas cisteínas do sensor de Keap1 (varas azuis), o que conduce a un cambio conformacional e unha actividade de adaptador substrato prexudicada. O Keap1 libre non é rexenerado, eo Nrf2 recén sintetizado acumúlase e se transloca ao núcleo.
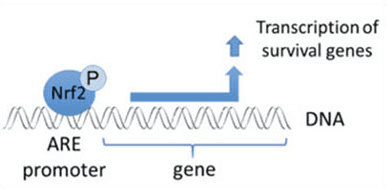
Ademais de servir como proteína adaptadora de sustrato de ligase de ubiquitina, Keap1 tamén é o sensor dunha ampla gama de activadores de pequenas moléculas de Nrf2 (inductores denominados) [9]. Os indutores bloquean o ciclo da degradación mediada por KeapXUMX de Nrf1 modificando químicamente residuos de cisteína específicos dentro de Keap2 [1], [10] ou interrompendo directamente a interfaz de conexión Keap11: Nrf1 [2], [12]. En consecuencia, Nrf13 non se degrada e o factor de transcrición acumúlase e se transloca ao núcleo (Fig. 2B), onde forma un heterodímero cunha pequena proteína Maf; liga aos elementos de resposta antioxidante, as regras reguladoras de arriba dos seus xenes obxecto de aprendizaxe; e inicia a transcrición [1], [14], [15]. A batería de obxectivos Nrf16 comprende proteínas con diversas funcións citoprotectoras, incluíndo encimas de metabolismo xenobiótico, proteínas con funcións antioxidantes e antiinflamatorias e subunidades proteasómicas, así como proteínas que regulan a homeostase redox celular e participan no metabolismo intermediario.
Nrf2: un regulador principal da homeostasis redox celular
A función de Nrf2 como regulador principal da homeostase redox celular é amplamente recoñecida. A expresión xénica das subunidades tanto catalíticas como reguladoras da β-glutamil cisteína ligase, o encima que cataliza o paso limitante da velocidade na biosíntese do glutatión reducido (GSH), está directamente regulada por Nrf2 [17]. A subunidade xCT do sistema xc-, que importa cistina ás células, tamén é un obxectivo de transcrición directa de Nrf2 [18]. Na célula, a cistina convértese en cisteína, un precursor da biosíntese de GSH. Ademais do seu papel na biosíntese de GSH, Nrf2 proporciona os medios para o mantemento do glutatión no seu estado reducido mediante a regulación transcripcional coordinada da glutatión redutase 1 [19], [20], que reduce o glutatión oxidado a GSH usando equivalentes reductores de NADPH. . O NADPH necesario é proporcionado por catro encimas xeradores de NADPH principais, o encima málico 1 (ME1), isocitrato deshidroxenase 1 (IDH1), glicosa-6-fosfato deshidroxenase (G6PD) e 6-fosfogluconato deshidroxenase (PGD), todos eles. regulada transcripcionalmente en parte por Nrf2 (Fig. 2) [21], [22], [23], [24]. Curiosamente, Nrf2 tamén regula a expresión xenética inducible das formas citosólicas, microsómicas e mitocondriais da aldehído deshidroxenase [25], que usan NAD(P)+ como cofactor, dando lugar a NAD(P)H. De feito, os niveis de NADPH e a relación NADPH/NADP+ son máis baixos nos fibroblastos embrionarios illados de ratos Nrf2-knockout (Nrf2-KO) en comparación coas células dos seus homólogos de tipo salvaxe (WT), e os niveis de NADPH diminúen ao derrubar Nrf2 en ratos. liñas celulares cancerosas con Nrf2 constitutivamente activo [26]. Como era de esperar, os niveis de GSH son máis baixos nas células nas que se interrompeu o Nrf2; pola contra, a activación de Nrf2 por medios xenéticos ou farmacolóxicos leva á regulación positiva de GSH [27], [28], [29]. É importante destacar que Nrf2 tamén regula a expresión xénica da tiorredoxina [30], [31], [32], a tioredoxina reductase 1 [28], [29], [32], [33] e a sulfiredoxina [34], que son esenciais. para a redución de tioles proteicos oxidados.
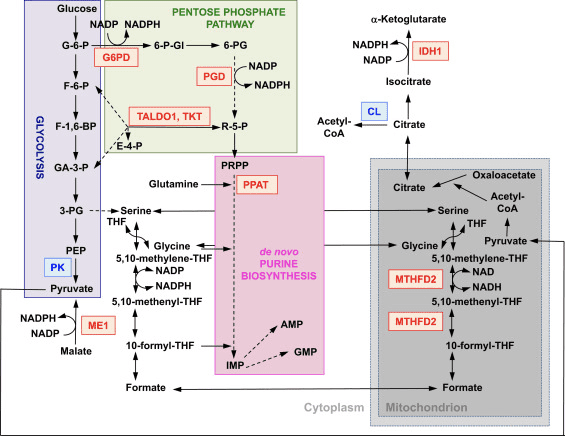
Figura 2 O papel de Nrf2 no metabolismo das células que proliferan rápidamente. Nrf2 é un regulador positivo dos xenes que codifican encimas tanto no brazo oxidativo [é dicir, glicosa-6-fosfato deshidroxenase (G6PD) e 6-fosfogluconato deshidroxenase (PGD)] como no brazo non oxidativo [é dicir, transaldolase 1 (TALDO1) e transcetolase. TKT)] da vía da pentosa fosfato. G6PD e PGD xeran NADPH. Nrf2 tamén regula a expresión xénica dos outros dous encimas xeradores de NADPH, o encima málico 1 (ME1) e a isocitrato deshidroxenase 1 (IDH1). A expresión xénica da fosforibosil pirofosfato amidotransferase (PPAT), que cataliza a entrada na vía biosintética das purinas de novo, tamén está regulada positivamente por Nrf2, así como a expresión da metilentetrahidrofolato deshidroxenase 2 (MTHFD2), un encima mitocondrial crítico con proporcionando unidades dun carbono para a biosíntese de purinas de novo. A piruvato quinase (PK) está regulada negativamente por Nrf2 e espérase que favoreza a acumulación de intermediarios glicolíticos e, xunto coa G6PD, a canalización de metabolitos a través da vía da pentosa fosfato e a síntese de ácidos nucleicos, aminoácidos e fosfolípidos. Nrf2 regula negativamente a expresión xénica da ATP-citrato liase (CL), o que pode aumentar a dispoñibilidade de citrato para a utilización mitocondrial ou (a través do isocitrato) para IDH1. O vermello e o azul indican regulación positiva e negativa, respectivamente. A mitocondria móstrase en gris. Abreviaturas de metabolitos: G-6-P, glicosa 6-fosfato; F-6-P, frutosa 6-fosfato; F-1,6-BP, frutosa 1,6-bisfosfato; GA-3-P, gliceraldehido 3-fosfato; 3-PG, 3-fosfoglicerato; PEP, fosfoenolpiruvato; 6-P-Gl, 6-fosfogluconolactona; 6-PG, 6-fosfogluconato; R-5-P, ribulosa 5-fosfato; PRPP, 5-fosforibosil-a-1-pirofosfato; THF, tetrahidrofolato; IMP, monofosfato de inosina; AMP, monofosfato de adenosina; GMP, monofosfato de guanosina.
Dado o papel crucial de Nrf2 como un regulador principal da homeostase redox móbil, non é sorprendente que, en comparación coas células WT, os niveis de especies reactivas de osíxeno (ROS) sexan máis altas nas células nas que Nrf2 foi interrompido (Nrf2-KO) [35]. Esta diferenza é especialmente rechamante ante os axentes que causan estrés oxidativo. Ademais, as células deficientes en Nrf2 son moito máis sensibles á toxicidade dos oxidantes de varios tipos e non poden ser protexidas polos inductores Nrf2, que, nas mesmas condicións, proporcionan unha protección eficiente e duradeira ás células WT [29], [36] , [37]. Ademais da homeostase redox móbil global, Nrf2 tamén é crítico para o mantemento da homeostase redox mitocondrial. Deste xeito, en comparación con WT, o total da piscina NADH mitocondrial aumenta significativamente en Keap1-KO e diminuíu drasticamente nas células Nrf2-KO [35].
Usando imaxes de células vivas, seguimos monitores das taxas de produción de ROS en coculturas glioneuronales primarias e rodais de tecido cerebral illados de ratones WT, Nrf2-KO ou Keap1-knockdown (Keap1-KD) [38]. Como era de esperar, a taxa de produción de ROS foi máis rápida en células e tecidos Nrf2-KO en comparación coas súas contrapartes WT. Con todo, fixemos a observación inesperada de que, en comparación con WT, as células Keap1-KD tamén teñen maiores taxas de produción de ROS, aínda que a magnitude da diferenza entre os genotipos WT e Keap1-KD era menor que a entre WT e Nrf2-KO . A continuación, analizáronse os niveis de mRNA de NOX2 e NOX4, as subunidades catalíticas das dúas isoformas NADPH oxidasa (NOX) implicadas na patoloxía do cerebro e descubriron que NOX2 aumentou drasticamente en condicións de deficiencia de Nrf2, mentres que NOX4 está regulamentado cando Nrf2 Está activamente constituída, aínda que en menor medida. Cuantitativamente, a magnitude do upregulation en células e tecidos dos ratones mutantes compara os aumentos correspondentes na produción de ROS [38]. Curiosamente, non só Nrf2 regula NADPH oxidasa, pero o ROS producido por NADPH oxidasa pode activar Nrf2, como se mostra nas células epiteliais pulmonares e cardiomiocitos [39], [40]. Ademais, un estudo moi recente demostrou que a activación dependente de NADPH oxidasa de Nrf2 constitúe un importante mecanismo endóxeno para a protección contra o dano mitocondrial e a morte celular no corazón durante a sobrecarga crónica de presión [41].
Ademais da actividade catalítica de NADPH oxidasa, a respiración mitocondrial é outra importante fonte intracelular de ROS. Mediante o uso da sonda MitoSOX específica da mitocondria, examinamos a contribución de ROS de orixe mitocondrial á produción total de ROS en cogumelos glioneuronales primarios illados de ratones WT, Nrf2-KO ou Keap1-KD [38]. Como era de esperar, as células Nrf2-KO tiñan taxas máis elevadas de produción de mitocondria ROS que WT. De acordo cos resultados da produción global de ROS, as taxas de produción de mitocondria ROS en Keap1-KD tamén foron máis altas en comparación coas células WT. Importante: o bloqueo complexo I con rotenona causou un aumento dramático na produción de ROS mitocondrial nas células WT e Keap1-KD, pero non tivo efecto nas células Nrf2-KO. En contraste co aumento esperado da produción de ROS mitocondrial nas células WT despois da adición de piruvato (para mellorar a dispoñibilidade de NADH, aumentar o potencial de membrana mitocondrial e normalizar a respiración), a produción de ROS diminuíu nas células Nrf2-KO. Xuntos, estes resultados suxiren fortemente que, a falta de Nrf2: (i) a actividade do complexo I está prexudicada, (ii) a actividade prexudicada do complexo I é debido á limitación de sustratos, e (iii) a actividade alterada do complexo Eu son un dos motivos principais da produción de ROS mitocondrial, posiblemente debido ao fluxo de electróns inversos do complexo II.
Nrf2 afecta o potencial de membrana mitocondrial e a respiración
O potencial de membrana mitocondrial (??m) é un indicador universal da saúde mitocondrial e do estado metabólico da célula. Nunha célula sa, ??m é mantido pola cadea respiratoria mitocondrial. Curiosamente, un etiquetaxe isotópico estable con aminoácidos nun estudo proteómico baseado en cultivos na liña celular MCF10A do epitelio de mama humano non tumorixeno negativo do receptor de estróxenos demostrou que o compoñente da cadea de transporte de electróns mitocondriais NDUFA4 está regulado pola activación farmacolóxica (por sulforafano) de Nrf2. mentres que a regulación xenética de Nrf2 (por derrubamento de Keap1) leva á baixada das subunidades da citocromo c oxidase COX2 e COX4I1 [42]. Un estudo do proteoma hepático mediante electroforese en xel bidimensional e espectrometría de masas de desorción/ionización láser asistida por matriz descubriu que Nrf2 regula a expresión da subunidade ATP sintase? [43]. Ademais, a proteína mitocondrial DJ-1, que xoga un papel no mantemento da actividade do complexo I [44], estabiliza Nrf2 [45], [46], aínda que os efectos neuroprotectores da activación farmacolóxica ou xenética. de Nrf2 son independentes de DJ-1 [47]. Non obstante, non se investigaron as consecuencias destas observacións para a función mitocondrial.
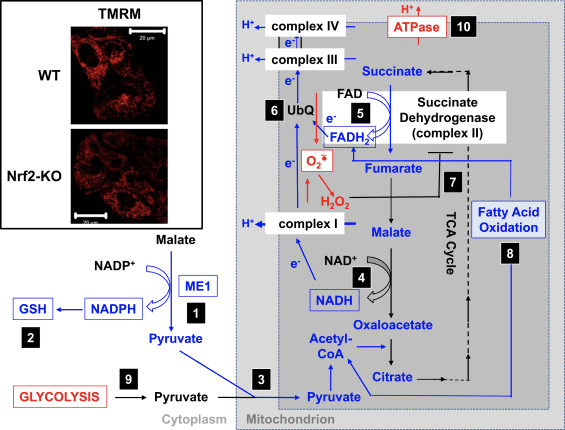
De acordo coa actividade prexudicada do complexo I en condicións de deficiencia de Nrf2, o ??m basal é menor nos fibroblastos embrionarios de rato (MEF) Nrf2-KO e nas células glioneuronais primarias cultivadas en comparación coas súas contrapartes WT (Fig. 3, recuadro). [35]. Pola contra, o ??m basal é maior cando Nrf2 está xeneticamente regulado de forma constitutiva (por derrubamento ou eliminación de Keap1). Estas diferenzas en ??m entre os xenotipos indican que a respiración está afectada pola actividade de Nrf2. De feito, a avaliación do consumo de osíxeno no estado basal revelou que, en comparación co WT, o consumo de osíxeno é menor nos MEF Nrf2-KO e Keap1-KO, nun ~50 e ~35%, respectivamente.
Figura 3 Mecanismo proposto para a función mitocondrial comprometida baixo condicións de deficiencia de Nrf2. (1) A diminución dos niveis de ME1, IDH1, G6PD e PGD resulta en niveis máis baixos de NADPH. (2) Os niveis de GSH tamén son baixos. (3) A baixa actividade de ME1 pode diminuír a piscina de piruvato que entra nas mitocondrias. (4) A xeración de NADH é máis lenta, o que provoca unha diminución da actividade do complexo I e un aumento da produción de ROS mitocondrial. (5) Tamén diminúe a redución de FAD a FADH2 nas proteínas mitocondriais, diminuíndo o fluxo de electróns de FADH2 a UbQ e ao complexo III. (6) A formación máis lenta de UbQH2 pode diminuír a actividade enzimática da succinato deshidroxenase. (7) O aumento dos niveis de ROS pode inhibir aínda máis a actividade do complexo II. (8) A menor eficiencia da oxidación dos ácidos graxos contribúe á diminución da dispoñibilidade de substrato para a respiración mitocondrial. (9) A glicólise mellora como mecanismo compensatorio para a diminución da produción de ATP na fosforilación oxidativa. (10) A ATP sintase opera á inversa para manter ??m. O vermello e o azul indican unha regulación positiva e unha regulación negativa, respectivamente. As caixas indican a dispoñibilidade de probas experimentais. O recuadro mostra imaxes de mitocondrias de astrocitos corticais WT e Nrf2-KO visualizadas pola sonda fluorescente potenciométrica de éster metílico de tetrametilrodamina (TMRM; 25 nM). Barra de escala, 20 �m.
Estas diferenzas en ??m e respiración entre os xenotipos reflíctese pola taxa de utilización dos substratos para a respiración mitocondrial. A aplicación de substratos para o ciclo do ácido tricarboxílico (TCA) (malato/piruvato, que á súa vez aumentan a produción do substrato do complexo I NADH) ou succinato de metilo, un substrato do complexo II, provoca un aumento gradual de ??m en ambos os dous WT. e as neuronas Keap1-KD, pero a taxa de aumento é maior nas células Keap1-KD. Máis importante aínda, as formas da resposta a estes substratos do ciclo de TCA son diferentes entre os dous xenotipos, polo que o rápido aumento de ??m nas células Keap1-KD tras a adición de substrato é seguido por unha caída rápida en lugar dunha meseta, o que suxire unha consumo rápido de substrato. Estes achados están en estreita concordancia cos niveis moito máis baixos (un 50 %) de malato, piruvato e succinato que se observaron despois dun pulso de 70 h de [U-1C13]glicosa en Keap6-KO en comparación co WT MEF. células [1]. Nas neuronas Nrf24-KO, só o piruvato é capaz de aumentar o ??m, mentres que o malato e o succinato de metilo provocan unha leve despolarización. O efecto de Nrf2 na produción de substrato mitocondrial parece ser o principal mecanismo polo cal Nrf2 afecta a función mitocondrial. O índice redox de NADH mitocondrial (o equilibrio entre o consumo de NADH polo complexo I e a produción de NADPH no ciclo TCA) é significativamente menor nas células Nrf2-KO en comparación coas súas contrapartes WT e, ademais, as taxas de rexeneración dos grupos de NADH e FADH2 despois da inhibición do complexo IV (mediante o uso de NaCN) son máis lentos nas células mutantes.
Nas mitocondrias illadas do cerebro e do fígado murinos, a suplementación de substratos para o complexo I ou para o complexo II aumenta a taxa de consumo de osíxeno con máis forza cando se activa Nrf2 e de forma menos eficiente cando se interrompe [2]. Así, o malato induce unha maior taxa de consumo de osíxeno en Keap35-KD en comparación co WT, pero o seu efecto é máis débil nas mitocondrias Nrf1-KO. Do mesmo xeito, en presenza de rotenona (cando se inhibe o complexo I), o succinato activa o consumo de osíxeno en maior medida en Keap2-KD en comparación co WT, mentres que a resposta nas mitocondrias Nrf1-KO diminúe. Ademais, os cultivos neuronais primarios Nrf2-KO e os ratos son máis sensibles á toxicidade dos inhibidores do complexo II ácido 2-nitropropiónico e malonato, mentres que o transplante intraestriatal de astrocitos que sobreexpresan Nrf3 é protector [2], [48]. Do mesmo xeito, os ratos Nrf49-KO son máis sensibles, mentres que a activación xenética ou farmacolóxica de Nrf2 ten efectos protectores contra a neurotoxicidade causada polo inhibidor do complexo I do ión 2-metil-1-fenilpiridinio no 4-metil-1-fenil-4. Modelo animal de 1,2,3,6-tetrahidropiridina da enfermidade de Parkinson [49], [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61].
A relación de control respiratorio (RCR), a proporción do estado 3 (estimulado por ADP) e a respiración do estado 4 (sen ADP presente), diminúe en ausencia de Nrf2, pero o RCR é similar entre as mitocondrias Keap1-KD e WT [35]. ]. Como o RCR é unha indicación do grao de acoplamento da actividade da cadea respiratoria mitocondrial á fosforilación oxidativa, este achado indica que a maior taxa de respiración nas mitocondrias Keap1-KD non se debe ao desacoplamento da fosforilación oxidativa. Ademais suxire que a fosforilación oxidativa é máis eficiente cando se activa Nrf2. A maior taxa de respiración nas mitocondrias Keap1-KD é consistente cos niveis máis altos de produción de ROS mitocondrial [38] xa que as taxas de respiración máis altas poden provocar un aumento da fuga de electróns. Non obstante, en condicións de estrés oxidativo, o aumento da produción de ROS vese contrarrestado pola regulación transcripcional da proteína de desacoplamento 2 (UCP3) dependente de Nrf3, que aumenta a condutancia de protóns da membrana interna mitocondrial e, en consecuencia, diminúe a produción de superóxido [62]. Moi recentemente, demostrouse que o produto da peroxidación lipídica 4-hidroxi-2-nonenal media na regulación positiva de UCP2 dependente de Nrf3 nos cardiomiocitos; isto pode ser particularmente importante para a protección en condicións de estrés oxidativo como as durante a isquemia-reperfusión [63].
Nrf2 afecta a eficacia da fosforilación oxidativa ea síntese de ATP
De acordo co efecto de Nrf2 sobre a respiración, nas mitocondrias do cerebro e do fígado, a deficiencia de Nrf2 ten como resultado unha diminución da eficiencia da fosforilación oxidativa (según o estimado pola relación de ADP e osíxeno, que se consume para a síntese de ATP), mentres que a activación de Nrf2 (Keap1). -KD) ten o efecto contrario [35]. En comparación co WT, os niveis de ATP son significativamente máis altos nas células con regulación positiva constitutiva de Nrf2 e máis baixos cando Nrf2 é derrubado [64] ou interrompido [35]. Ademais, o uso de inhibidores da fosforilación oxidativa (oligomicina) ou da glicólise (ácido iodoacético) revelou que Nrf2 cambia a forma en que as células producen ATP. Así, nas neuronas WT, a oligomicina provoca unha caída completa do ATP e o ácido iodoacético non ten ningún efecto adicional. Sorprendentemente, nas células Nrf2-KO, a oligomicina aumenta os niveis de ATP, que despois son lentamente, pero completamente, esgotados polo ácido iodoacético, o que indica que, en ausencia de Nrf2, a glicólise, e non a fosforilación oxidativa, é a principal fonte de produción de ATP. Curiosamente, a pesar da maior eficiencia da fosforilación oxidativa nas células Keap1-KD, a adición de oligomicina produce unha diminución de ~80% nos niveis de ATP, e o ácido iodoacético provoca unha diminución adicional de ~20%. Así, a deficiencia de Nrf2 ou a súa activación constitutiva reduce a contribución da fosforilación oxidativa e aumenta a contribución da glicólise á síntese de ATP. Este efecto é particularmente pronunciado cando Nrf2 está ausente e é consistente coa dependencia do ??m da presenza de glicosa no medio [35] e dos niveis incrementados de intermediarios glicolíticos (G-6-P, F-6-P). , fosfato de dihidroxiacetona, piruvato e lactato) despois da caída de Nrf2 [24].
O aumento dos niveis de ATP despois da inhibición da F1F0-ATPase pola oligomicina indica que, en ausencia de Nrf2, a F1F0-ATPase funciona como ATPase e non como ATP sintase, é dicir, funciona ao revés. Tal reversión da actividade probablemente reflicta a necesidade de bombear protóns a través da membrana mitocondrial interna nun intento de manter o ??m, que é crucial para a integridade funcional deste orgánulo. A reversión da función da F1F0-ATPase tamén se evidencia pola despolarización mitocondrial observada tras a administración de oligomicina ás células Nrf2-KO, o que contrasta marcadamente coa hiperpolarización que ocorre nas súas contrapartes con deficiencia de WT ou Keap1 [35]. En xeral, parece que en condicións de deficiencia de Nrf2 o ATP prodúcese principalmente na glicólise, e este ATP é entón usado en parte pola F1F0-ATPase para manter o ??m.
Nrf2 mellora a oxidación de ácidos graxos mitocondriales
O efecto da deficiencia de Nrf2 sobre o ??m é particularmente pronunciado cando as células se incuban en medio sen glicosa, e o ??m é un 50% menor en Nrf2-KO en comparación coas células WT [35]. En condicións de privación de glicosa, a oxidación de ácidos graxos mitocondriais (FAO) é un importante provedor de substratos para a respiración e a fosforilación oxidativa, o que suxire que Nrf2 pode afectar á FAO. De feito, a eficiencia da FAO tanto para o ácido palmítico de ácidos graxos saturados de cadea longa (C16:0) como para o ácido hexanoico de cadea curta (C6:0) é maior nos MEF Keap1-KO e nas mitocondrias cardíacas e hepáticas illadas que nos seus. homólogos WT, mentres que é máis baixo nas células Nrf2-KO e nas mitocondrias [65]. Estes efectos tamén son moi relevantes para os seres humanos: de feito, os cambios metabólicos indicativos dunha mellor integración da FAO coa actividade do ciclo TCA foron reportados en estudos de intervención humana con dietas ricas en glucorafanina, o precursor do clásico activador Nrf2 sulforafano. 66].
Durante o primeiro paso da FAO mitocondrial, o hidróxeno pro-R do carbono β sae como un hidruro que reduce o cofactor FAD a FADH2, que á súa vez transfire electróns á ubiquinona (UbQ) na cadea respiratoria, contribuíndo finalmente á produción de ATP. . Mentres que a estimulación da FAO por palmitoilcarnitina en ausencia de glicosa provoca o aumento esperado dos niveis de ATP nas células WT e Keap1-KO, sendo o aumento de ATP máis rápido nas células Keap1-KO, o tratamento idéntico non produce cambios de ATP en Nrf2-KO. MEF [65]. Este experimento demostra que, en ausencia de Nrf2, a FAO está suprimida e, ademais, implica a supresión da FAO como unha das razóns dos niveis máis baixos de ATP en condicións de deficiencia de Nrf2 [35], [64].
Notablemente, as células T 293 humanas nas cales Nrf2 foi silenciada teñen unha menor expresión de CPT1 e CPT2 [67], dúas isoformas de carnitina palmitoyltransferase (CPT), o enzima que limita a velocidade na FAO mitocondrial. De acordo, os niveis de mRNA de Cpt1 son máis baixos nos fígados de Nrf2-KO en comparación cos ratones WT [68]. O CPT cataliza a transferencia do grupo acilo dun acil-CoA graxo de cadea longa a partir da coenzima A a l-carnitina e permite a importación de acilcarnitina a partir do citoplasma nas mitocondrias. Aínda que isto non foi examinado ata a data, é posible que, ademais dos efectos transcripcionais sobre a expresión CPT1, Nrf2 tamén pode afectar a función desta enzima controlando os niveis do seu principal inhibidor alostérico, malonyl-CoA. Isto ocorre porque, por un mecanismo que actualmente non está claro, Nrf2 regula negativamente a expresión de estearoílo CoA desaturase (SCD) [69] e citrato liase (CL) [69], [70]. Curiosamente, a eliminación ou a inhibición da SCD orixina unha maior fosforilación e activación da proteína quinasa AMP (AMPK) [71], [72], [73] e pódese especular que, a falta de Nrf2, os niveis de SCD aumentará, ao mesmo tempo baixará a actividade de AMPK. Isto podería ser aínda máis composto polos reducidos niveis de proteína de AMPK que se observaron nos fígados dos ratones Nrf2-KO [68], un achado que está en estrema acordo cos niveis aumentados de AMPK, que se informaron nos fígados de Keap1-KD ratos [74]. Unha consecuencia da diminución da actividade de AMPK é o alivio da súa fosforilación inhibitiva (en Ser79) de acetil-CoA carboxilase (ACC) [75], que pode ser regulada regulamentariamente de forma transcrita na ausencia de Nrf2 porque está regulada pola activación Nrf2 [70 ]. A elevada actividade ACC, en combinación coa expresión CL regulada que aumenta a produción de acetil-CoA, substrato para ACC, pode aumentar os niveis do produto ACC, malonyl-CoA. Os altos niveis de malonil-CoA inhiben o CPT, diminuíndo así o transporte de ácidos graxos na mitocondria. Finalmente, Nrf2 regula positivamente a expresión de CD36 [76], unha translocase que importa ácidos graxos en plasma e membranas mitocondriais. Así, un mecanismo polo que Nrf2 pode afectar a eficiencia da FAO mitocondrial é a regulación da importación de ácidos graxos de cadea longa na mitocondria.
Ademais da regulación transcripcional directa, Nrf2 tamén pode alterar a eficacia da FAO mitocondrial polos seus efectos sobre o metabolismo redox celular. Isto pode ser especialmente relevante cando a actividade Nrf2 é baixa ou ausente, as condicións que cambian o estado redox móbil cara ao estado oxidado. De feito, varias enzimas da FAO foron identificadas como sensibles aos cambios redox. Unha delas é acil-CoA deshidroxenase (VLCAD) de moi cadea longa, que aporta máis de 80% á actividade de deshidrogenación de palmitoil-CoA nos tecidos humanos [77]. Curiosamente, Hurd et al. [78] demostraron que VLCAD contén residuos de cisteína que cambian significativamente o seu estado redox despois da exposición de mitocondrias de corazón de rata illadas a H2O2. Ademais, a S-nitrosilación do VLCAD hepático murino en Cys238 mellora a eficiencia catalítica da enzima [79], e é probable que a oxidación da mesma cisteína poida ter o efecto contrario, reducindo finalmente a eficiencia da FAO mitocondrial. Polo tanto, é posible que, aínda que os niveis de expresión de VLCAD non sexan significativamente diferentes en WT, Nrf2-KO ou Keap1-KO MEF [65], a actividade enzimática de VLCAD podería ser menor en ausencia de Nrf2 debido aos niveis máis altos de ROS.
En base a todos estes achados, pódese propoñer que (Fig. 3): en ausencia de Nrf2, os niveis de NADPH son máis baixos debido á diminución da expresión de ME1, IDH1, G6PD e PGD. Os niveis de glutatión reducido tamén son máis baixos debido á diminución da expresión dos encimas que participan na súa biosíntese e rexeneración e aos niveis máis baixos de NADPH que son necesarios para a conversión do glutatión oxidado á forma reducida. A baixa expresión de ME1 diminuirá a piscina de piruvato que entra nas mitocondrias, e a glicólise se converte na principal fonte de piruvato. A xeración de NADH é máis lenta, o que provoca unha diminución da actividade do complexo I e un aumento da produción de ROS mitocondrial. A redución de FAD a FADH2 tamén é máis lenta, polo menos en parte debido a unha oxidación de ácidos graxos menos eficiente, comprometendo o fluxo de electróns de FADH2 a UbQ e ao complexo III. Como UbQH2 é un activador da succinato deshidroxenase [80], a ralentización da súa formación pode diminuír a actividade enzimática da succinato deshidroxenase. O aumento dos niveis de superóxido e peróxido de hidróxeno pode inhibir aínda máis a actividade do complexo II [81]. A menor eficiencia da oxidación dos ácidos graxos contribúe á diminución da dispoñibilidade de substrato para a respiración mitocondrial e a produción de ATP na fosforilación oxidativa. Como mecanismo compensatorio, mellora a glicólise. A ATP sintase funciona á inversa, como unha ATPasa, nun intento de manter o ??m.
Nrf2 e Biogénese mitocondrial
Informeuse de que, en comparación co WT, os fígados dos ratos Nrf2-KO teñen un contido mitocondrial máis baixo (según se determina pola proporción de ADN mitocondrial e nuclear); isto diminúe aínda máis nun rápido de 24 h tanto en ratos WT como Nrf2-KO; pola contra, aínda que non é diferente do WT en condicións normais de alimentación, o contido mitocondrial en ratos con alta actividade de Nrf2 non se ve afectado polo xaxún [82]. Curiosamente, a suplementación co ácido activador Nrf2 (R)-?-lipoico [83], [84], [85] promove a bioxénese mitocondrial nos adipocitos 3T3-L1 [86]. Dúas clases de reguladores de transcrición nucleares desempeñan un papel crítico na bioxénese mitocondrial. A primeira clase son os factores de transcrición, como os factores respiratorios nucleares11 e 2, que controlan a expresión dos xenes que codifican as subunidades dos cinco complexos respiratorios, os compoñentes de tradución mitocondriais e os encimas biosintéticos hemo que están localizados na matriz mitocondrial [88]. Piantadosi et al. [89] demostraron que a subregulación transcripcional dependente de Nrf2 do factor respiratorio nuclear 1 promove a bioxénese mitocondrial e protexe contra a citotoxicidade do axente quimioterapéutico cardiotóxico antraciclina doxorrubicina. Pola contra, Zhang et al. [82] informaron de que a activación xenética de Nrf2 non afecta a expresión de ARNm basal do factor respiratorio nuclear 1 no fígado murino.
A segunda clase de reguladores transcripcionais nucleares con funcións críticas na bioxénese mitocondrial son os coactivadores transcripcionais, como o receptor activado polo proliferador de peroxisomas ? coactivadores (PGC)1? e 1?, que interactúan con factores de transcrición, a maquinaria basal de transcrición e de empalme de ARN e encimas modificadoras de histonas [88], [90], [91]. A expresión da familia de coactivadores PGC1 está influenciada por numerosos sinais ambientais. O tratamento de fibroblastos humanos co sulforafano activador Nrf2 provoca un aumento da masa mitocondrial e a indución de PGC1? e PGC1? [92], aínda que neste estudo non se examinou a potencial dependencia de Nrf2. Non obstante, os ratos diabéticos nos que Nrf2 é activado pola caída hipomórfica do xene Keap1 (db/db:Keap1flox/?:Nrf2+/+) ou interrompido (db/db:Keap1flox/?:Nrf2?/?) teñen PGC1 hepático máis baixo? niveis de expresión que os animais control (db/db:Keap1flox/+:Nrf2+/+) [93]. Non hai diferenzas nos niveis de ARNm para PGC1? obsérvanse en fígados de ratos non diabéticos que son WT ou Nrf2-KO, mentres que estes niveis son máis baixos nos animais que sobreexpresan Nrf2 (Keap1-KD e Keap1-KO específico do fígado) [82]. En particular, un rápido de 24 horas aumenta os niveis de PGC1? ARNm nos fígados de ratos de todos os xenotipos, pero o aumento é significativamente maior nos fígados de Nrf2-KO en comparación cos ratos que sobreexpresan WT ou Nrf2. En comparación co WT, os ratos Nrf2-KO que experimentan infección séptica ou lesión pulmonar aguda debido á infección mostran unha regulación positiva transcripcional atenuada do factor respiratorio nuclear 1 e PGC1? [94], [95]. En conxunto, estas observacións suxiren que o papel de Nrf2 no mantemento dos niveis de factor respiratorio nuclear 1 e PGC1? é complexo e faise máis destacado en condicións de estrés.
Ademais da expresión dos xenes que codifican proteínas mitocondriais, a bioxénese mitocondrial require a síntese de nucleótidos. A activación xenética de Nrf2 mellora a biosíntese de purinas regulando a vía da pentosa fosfato e o metabolismo de folato e glutamina, especialmente nas células que proliferan rapidamente (Fig. 2) [24]. A análise do transcriptoma de Drosophila mutante deficiente para a serina/treonina proteína quinase mitocondrial inducida por PTEN quinase 1 (PINK1) mostrou que a disfunción mitocondrial leva á regulación positiva da transcrición dos xenes que afectan o metabolismo dos nucleótidos [96], o que suxire que a bionucleótida mellora representa un mecanismo de protección contra as consecuencias neurotóxicas da deficiencia de PINK1. Nrf2 regula a expresión da fosforibosil pirofosfato amidotransferase (PPAT), que cataliza a entrada na vía biosintética de nucleótidos purinos de novo e a metilentetrahidrofolato deshidroxenase mitocondrial 2 (MTHFD2) (Fig. 2). Este último é un encima bifuncional con actividades deshidroxenase e ciclohidrolasa que é fundamental para proporcionar tanto glicina como formiato como fontes de unidades dun carbono para a biosíntese de purinas en células de crecemento rápido [97]. Polo tanto, é probable que a activación de Nrf2 poida ser protectora e revertir a disfunción mitocondrial na deficiencia de PINK1. De feito, a activación farmacolóxica de Nrf2 polo sulforafano, ou o triterpenoide RTA-408, restaura ??m e protexe as células deficientes en PINK1 contra a toxicidade da dopamina [98]. Aínda que os mecanismos subxacentes parecen ser complexos, en conxunto, estes achados indican que a actividade de Nrf2 pode afectar á bioxénese mitocondrial ao influír nos niveis de expresión dos factores críticos de transcrición e dos coactivadores, así como ao mellorar a biosíntese de nucleótidos.
Nrf2 e integridade mitocondrial
Aínda que a evidencia directa non sempre está dispoñible, hai indicios fortes de que Nrf2 é importante para a integridade mitocondrial, especialmente en condicións de estrés oxidativo. As mitocondrias illadas do cerebro e do fígado das ratas que se administraron unha única dose do activador Nrf2 sulforaphane son resistentes á apertura do poro de transición de permeabilidade mitocondrial (mPTP) causado polo hidroperóxido tert-butil [99], [100] oxidante. O mPTP, un complexo que permite que a membrana interna mitocondrial sexa permeable a moléculas con masa ata 1500 Da, foi recentemente identificada para ser formada por dímeros da F0F1-ATP synthase [101]. A resistencia sulphaphane mediada pola apertura de mPTP correlaciona co aumento das defensas antioxidantes e os niveis de GSH mitocondrial, glutation Xoxifase 1, enzima máxica 3 e Thioredoxin 2 están regulados en fraccións mitocondriais illadas de animais tratados con sulapaphane [100].
O dano das proteínas mitocondriais e o deterioro da respiración causados polo produto electrofílico de peroxidación lipídica 4-hidroxi-2-nonenal atenuanse nas mitocondrias illadas da cortiza cerebral dos ratos tratados con sulforafano [102]. Nas células epiteliais renais de rata e nos riles, o sulforafano é protector contra a toxicidade inducida por cisplatino e gentamicina e a perda de ??m[103], [104]. Tamén se observou protección contra un panel de oxidantes (superóxido, peróxido de hidróxeno, peroxinitrito) e electrófilos (4-hidroxi-2-nonenal e acroleína) e un aumento das defensas antioxidantes mitocondriais no tratamento de células do músculo liso aórtico de rata con sulforafano [105]. ]. Nun modelo de lesión renal aguda inducida por contraste, demostrouse recentemente que o preacondicionamento isquémico dos membros ten efectos protectores, incluíndo a inhibición da apertura do mPTP e o inchazo mitocondrial, pola activación de Nrf2 como consecuencia da inhibición de GSK3? [106].
A mitofaxia, o proceso polo cal as mitocondrias disfuncionais son absorbidas selectivamente polos autofagosomas e entregadas aos lisosomas para ser degradadas e recicladas pola célula, é esencial para a homeostase mitocondrial [107], [108]. Aínda que non se estableceu ningunha relación causal entre Nrf2 e a mitofaxia, hai evidencias de que o factor de transcrición pode ser importante no control da calidade mitocondrial ao desempeñar un papel na mitofaxia. Isto pode ser especialmente importante en condicións de estrés oxidativo. Así, nun modelo de sepsis, os aumentos dos niveis do marcador do autofagosoma MAP1 da cadea lixeira 3-II (LC3-II) e da proteína de carga p62 ás 24 h despois da infección suprimen os Nrf2-KO en comparación cos ratos WT [109] . Recentemente descubriuse un inductor de mitofaxia de pequenas moléculas (chamado inductor de mitofaxia mediado por p62, PMI); este composto de 1,4-difenil-1,2,3-triazol foi deseñado orixinalmente como un activador de Nrf2 que interrompe a interacción do factor de transcrición con Keap1 [110]. Do mesmo xeito que as células nas que Nrf2 está xeneticamente regulado (Keap1-KD ou Keap1-KO), as células expostas a PMI teñen ??m en repouso máis altos. É importante destacar que o aumento da localización mitocondrial de LC3 que se observa despois do tratamento con PMI das células WT non se produce nas células Nrf2-KO, o que suxire a implicación de Nrf2.
Por último, a análise ultraestructural das seccións hepáticas revelou a presenza de mitocondrias inchadas con crista reducida e membranas interrompidas en hepatocitos de Nrf2-KO, pero non WT, ratos que se alimentaron cunha dieta alta en graxa durante semanas 24; En particular, estes fígados mostran unha evidencia clara do estrés oxidativo e da inflamación [68]. Pódese concluír que Nrf2 ten un papel fundamental no mantemento da integridade mitocondrial en condicións de estrés oxidativo e inflamatorio.
Sulforaphane e os seus efectos sobre o cancro, a mortalidade, o envellecemento, o cerebro e o comportamento, as enfermidades cardíacas e moito máis
Os isotiocianatos son algúns dos compostos vexetais máis importantes que pode obter na súa dieta. Neste video fago o caso máis completo para eles que se fixo. ¿A atención curta? Saltar ao teu tema favorito premendo un dos puntos de tempo a continuación. Cadro de cronograma completo a continuación.
Seccións clave:
00: 01: 14 - Cáncer e mortalidade
00: 19: 04 - Envellecemento
00: 26: 30 - Cerebro e comportamento
00: 38: 06 - Recapitalización final
00: 40: 27 - Dose
Cadro de tempo completo:
00: 00: 34 - Introdución de sulforaphane, un foco principal do vídeo.
00: 01: 14 - Consumo e redución de vexetais cruciferos na mortalidade por todas as causas.
00: 02: 12 - Risco de cancro de próstata.
00: 02: 23 - Risco de cancro de vejiga.
00: 02: 34 - Cáncer de pulmón en risco de fumadores.
00: 02: 48 - Risco de cancro de mama.
00: 03: 13 - hipotético: e se xa ten cancro? (intervencionista)
00: 03: 35 - Mecanismo plausible que conduce os datos asociativos de cancro e mortalidade.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Evidencia animal que mostra un forte efecto do extracto de brotes de brócoli no desenvolvemento de tumores vesicales en ratas.
00: 06: 06 - Efecto da suplementación directa de sulforaphane en pacientes con cancro de próstata.
00: 07: 09 - Bioacumulación de metabolitos de isotiocianato no tecido de mama real.
00: 08: 32 - Inhibición das células nais do cancro de mama.
00: 08: 53 - Lección de historia: as brassicas establecéronse con propiedades de saúde mesmo na Roma antiga.
00: 09: 16 - A capacidade de Sulforaphane para mellorar a excreción de carcinóxenos (benceno, acroleína).
00: 09: 51 - NRF2 como un cambio xenético a través de elementos de resposta antioxidante.
00: 10: 10 - Como a activación de NRF2 aumenta a excreción de carcinóxenos a través de glutatión-S-conjugados.
00: 10: 34 - As coles de Bruxelas aumentan a glutatión-S-transferasa e reducen o dano do ADN.
00: 11: 20 - A bebida de brote de brócoli aumenta a excreción de benceno por 61%.
00: 13: 31 - O homogeneio de brotes de brócoli aumenta as encimas antioxidantes nas vías aéreas superiores.
00: 15: 45 - Consumo de vexetais cruciferos e mortalidade cardíaca.
00: 16: 55 - O polbo de brócolis mellora os lípidos sanguíneos eo risco de enfermidade cardíaca en diabéticos tipo 2.
00: 19: 04 - Comezo da sección de envellecemento.
00: 19: 21 - A dieta enriquecida con Sulforaphane mellora a vida útil dos escaravellos de 15 a 30% (en certas condicións).
00: 20: 34 - Importancia da baixa inflamación por lonxevidade.
00: 22: 05 - As verduras cruciferas e os brotes de brócoli parecen reducir unha gran variedade de marcadores inflamatorios en humanos.
00: 23: 40 - Recapitalización media: cancro, seccións de envellecemento
00: 24: 14 - Os estudos do rato suxiren que o sulforaphano pode mellorar a función inmune adaptativa na vellez.
00: 25: 18 - Sulforaphane mellorou o crecemento do cabelo nun modelo de calvície de rato. Imaxe en 00: 26: 10.
00: 26: 30 - Comezo da sección do cerebro e do comportamento.
00: 27: 18 - Efecto do extracto de brotes de brócoli no autismo.
00: 27: 48 - Efecto da glucoraphanina na esquizofrenia.
00: 28: 17 - Inicio da discusión de depresión (mecanismo plausible e estudos).
00: 31: 21 - O estudo do rato usando 10 diferentes modelos de depresión inducida polo estrés mostran sulforaphane igualmente efectivo como a fluoxetina (prozac).
00: 32: 00 - O estudo mostra que a inxestión directa de glucoraphanina en ratos é igualmente eficaz na prevención da depresión do modelo de estrés da derrota social.
00: 33: 01 - Inicio da sección de neurodegeneración.
00: 33: 30 - Sulforaphane e enfermidade de Alzheimer.
00: 33: 44 - Sulforaphane e enfermidade de Parkinson.
00: 33: 51 - Sulforaphane e enfermidade de Hungtington.
00: 34: 13 - Sulforaphane aumenta as proteínas de choque térmico.
00: 34: 43 - Inicio da sección traumática de lesións cerebrais.
00: 35: 01 - Sulforaphane inxectado inmediatamente despois de que o TBI mellore a memoria (estudo do rato).
00: 35: 55 - Sulforaphane e plasticidade neuronal.
00: 36: 32 - Sulforaphane mellora a aprendizaxe en modelo de diabetes tipo II en ratos.
00: 37: 19 - Distrofia muscular sulforaphana e duxena.
00: 37: 44 - Inhibición da myostatina nas células satélite do músculo (in vitro).
00: 38: 06 - Recapitulación de última hora: mortalidade e cancro, danos no ADN, estrés oxidativo e inflamación, excreción de benceno, enfermidade cardiovascular, diabetes tipo II, efectos sobre o cerebro (depresión, autismo, esquizofrenia e neurodegeneración), vía NRF2.
00: 40: 27 - Pensamentos en descubrir unha dose de brotes de brócoli ou sulforaphane.
00: 41: 01 - Anécdotas sobre o xermelo na casa.
00: 43: 14 - Sobre as temperaturas de cocción e actividade sulforaphane.
00: 43: 45 - Conversión de bacterias gut de sulforaphane a partir de glucoraphanin.
00: 44: 24 - Os suplementos funcionan mellor cando se combinan con mirosinasa activa de vexetais.
00: 44: 56 - Técnicas de cociña e vexetais crucíferos.
00: 46: 06 - Isotiocianatos como goitrógenos.
Nrf2 é un factor de transcrición que desempeña un papel importante no sistema de defensa antioxidante móbil do corpo humano. O elemento antioxidante sensible, ou ARE, é un mecanismo regulador dos xenes. Moitos estudos de investigación demostraron que o Nrf2 ou o factor 2 relacionado con NF-E2 regula unha gran variedade de xenes controlados por ARE a través de varios tipos de células. NRF2 tamén se atopou para desempeñar un papel esencial na protección celular e anti-canceríxeno, o que demostra que Nrf2 pode ser un tratamento eficaz na xestión de enfermidades neurodegenerativas e cancros que se cre que están causadas polo estrés oxidativo. Dr. Alex Jimenez DC, CCST Insight
Observacións finais
Aínda que aínda quedan moitas preguntas, a evidencia experimental dispoñible indica claramente que Nrf2 é un xogador importante no mantemento da homeostase mitocondrial e da integridade estrutural. Este papel tórnase particularmente crítico en condicións de estrés oxidativo, electrofílico e inflamatorio cando a capacidade de subregular as respostas citoprotutoras mediadas por Nrf2 inflúe na saúde xeral e na supervivencia da célula e do organismo. O papel de Nrf2 na función mitocondrial representa outra capa dos amplos mecanismos citoprotectores orquestados por este factor de transcrición. Como moitas condicións patolóxicas humanas teñen estrés oxidativo, inflamación e disfunción mitocondrial como compoñentes esenciais da súa patoxenia, a activación farmacolóxica de Nrf2 é unha promesa de prevención e tratamento da enfermidade. A comprensión integral dos mecanismos precisos mediante os que Nrf2 afecta a función mitocondrial é esencial para o deseño racional de ensaios clínicos futuros e pode ofrecer novos biomarcadores para controlar a eficacia terapéutica.
O propósito do artigo anterior era discutir, así como demostrar, o papel emerxente de Nrf2 na función mitocondrial. Nrf2 ou Factor nuclear eritroide 2 factor relacionado, é un regulador emerxente da resistencia celular aos oxidantes que pode contribuír ao estrés oxidativo, afectando a función celular e levando ao desenvolvemento de toxicidade, enfermidades crónicas e mesmo cancro. Aínda que a produción de oxidantes no corpo humano pode servir para varios propósitos, incluíndo a división celular, a inflamación, a función inmune, a autofaxia e a resposta ao estrés, é esencial controlar a súa sobreprodución para evitar problemas de saúde. O alcance da nosa información limítase aos problemas de saúde da columna vertebral e quiroprácticos. Para discutir o tema, non dubide en preguntar ao Dr. Jiménez ou en contacto connosco en�915-850-0900 .
Discusión de tema adicional: "Dor de espalda aguda
Dor nas costasÉ unha das causas máis frecuentes de discapacidade e días perdidos no traballo en todo o mundo. A dor nas costas atribúese á segunda razón máis común para as visitas ao médico, só superada en número por infeccións respiratorias superiores. Aproximadamente o 80 por cento da poboación experimentará dor nas costas polo menos unha vez ao longo da súa vida. A columna vertebral é unha estrutura complexa composta por ósos, articulacións, ligamentos e músculos, entre outros tecidos brandos. Debido a isto, lesións e / ou condicións agravadas, comodiscos herniados, eventualmente pode provocar síntomas de dor nas costas. As lesións por deporte ou por accidente automovilístico adoitan ser a causa máis frecuente de dores nas costas, con todo, ás veces o movemento máis simple pode ter resultados dolorosos. Afortunadamente, opcións de tratamento alternativas, como o coidado quiropráctico, poden axudar a aliviar a dor nas costas mediante o uso de axustes espiñais e manipulacións manuais, mellorando finalmente o alivio da dor.
Nrf2 soporta a activación dun grupo de encimas e xenes antioxidantes e desintoxicantes que protexen o corpo humano dos efectos de problemas de saúde asociados co aumento dos niveis de estrés oxidativo, como a enfermidade de Alzheimer. Unha variedade de substancias naturais demostraron que activan a vía Nrf2, que pode axudar a xestionar os síntomas das enfermidades neurodexenerativas. O obxectivo do artigo seguinte é discutir o papel fundamental de Nrf2 causado pola inflamación crónica.
Abstracto
A inflamación é a característica máis común de moitas enfermidades e complicacións crónicas, mentres desempeña un papel crítico na carcinoxénese. Varios estudos demostraron que Nrf2 contribúe ao proceso antiinflamatorio orquestando o recrutamento de células inflamatorias e regulando a expresión xénica a través do elemento de resposta antioxidante (ARE). A vía de sinalización Keap1 (proteína asociada a ECH similar a Kelch)/Nrf2 (factor 2 relacionado con NF-E45 p2)/ARE regula principalmente a expresión dos xenes antiinflamatorios e inhibe a progresión da inflamación. Polo tanto, a identificación de novos fitoquímicos antiinflamatorios dependentes de Nrf2 converteuse nun punto clave no descubrimento de fármacos. Nesta revisión, discutimos os membros da vía de sinal Keap1/Nrf2/ARE e os seus xenes augas abaixo, os efectos desta vía en modelos animais de enfermidades inflamatorias e a interferencia coa vía NF-?B. Ademais tamén discutimos sobre a regulación do inflamasoma NLRP3 por Nrf2. Ademais disto, resumimos o escenario actual do desenvolvemento de fitoquímicos antiinflamatorios e outros que median na vía de sinalización Nrf2/ARE.
A inflamación é un proceso complexo que ocorre cando os tecidos están infectados ou feridos por estímulos nocivos como patóxenos, danos ou irritantes. As células inmunitarias, os vasos sanguíneos e os mediadores moleculares están implicados nesta resposta protectora [1]. A inflamación tamén é un fenómeno patolóxico asociado a unha variedade de estados de enfermidades inducidos principalmente por factores físicos, químicos, biolóxicos e psicolóxicos. O obxectivo da inflamación é limitar e eliminar as causas do dano celular, limpar e / ou absorber células e tecidos necróticos e iniciar a reparación do tecido. Distínguense dúas formas distintas de inflamación: aguda e crónica. A inflamación aguda é autolimitada e beneficiosa para o hóspede, pero a inflamación crónica prolongada é unha característica común de moitas enfermidades e complicacións crónicas. A infiltración directa de moitas células inmunolóxicas mononucleares como monocitos, macrófagos, linfocitos e células plasmáticas, así como a produción de citoquinas inflamatorias, orixinan unha inflamación crónica. Recoñécese que a inflamación crónica ten un papel crítico na carcinogénesis [2]. En xeral, as vías de sinalización pro e antiinflamatorias interactúan no proceso inflamatorio normal.
No proceso inflamatorio patolóxico, primeiro activan os mastocitos, monocitos, macrófagos, linfocitos e outras células inmunitarias. Despois, as células son recrutadas para o lugar da lesión, o que resulta na xeración de especies reactivas do osíxeno (ROS) que danan as macromoléculas, incluíndo o ADN. Ao mesmo tempo, estas células inflamatorias tamén producen grandes cantidades de mediadores inflamatorios como citocinas, quimiocinas e prostaglandinas. Estes mediadores recrutan aínda máis macrófagos en sitios localizados de inflamación e activan directamente múltiples fervenzas de transdución de sinais e factores de transcrición asociados á inflamación. As vías de sinalización NF-?B (factor nuclear kappa B), MAPK (proteína quinase activada por mitógenos) e JAK (xanus quinase)-STAT (transdutores de sinal e activadores da transcrición) están implicadas no desenvolvemento da vía clásica da inflamación. [3], [4], [5]. Estudos anteriores revelaron que o factor de transcrición Nrf2 (factor 2 relacionado con NF-E45 p2) regula a expresión de encimas desintoxicantes da fase II, incluíndo NADPH, NAD(P)H quinona oxidorreductase 1, glutatión peroxidase, ferritina, hemo osixenase-1 (HO). -1), e xenes antioxidantes que protexen as células de diversas lesións mediante os seus efectos antiinflamatorios, influíndo así no curso da enfermidade [6], [7], [8].
Tendo en conta estas conclusións notables, o desenvolvemento de medicamentos terapéuticos específicos para enfermidades inflamatorias a través de vías de sinalización atraeu moito interese nos últimos anos. Nesta revisión, resumimos a investigación sobre o fluxo de sinalización de Keap1 (proteína asociada a Kelch-ECH) / Nrf2 (NF-E2 p45 factor 2) / ARE (elemento de resposta antioxidante).
Estrutura e regulación de Nrf2
Regulación Nrf1 dependente de Keap2
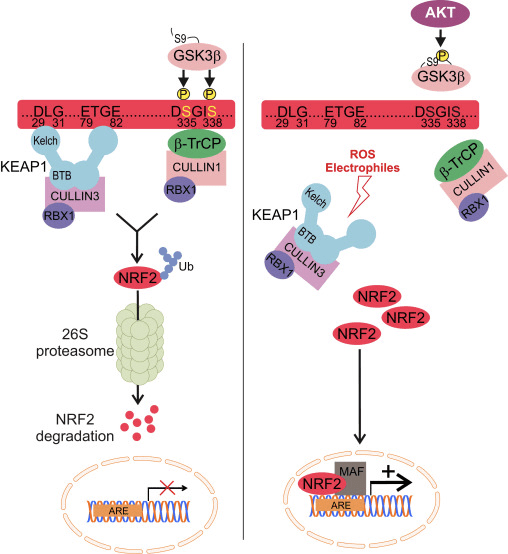
Nrf2 pertence á subfamilia Cap �n� Collar (CNC) e comprende sete dominios funcionais, Neh (homoloxía Nrf2-ECH) 1 a Neh7 [9], [10]. Neh1 é un dominio CNC-bZIP que permite a Nrf2 heterodimerizarse con pequenas proteínas do fibrosarcoma musculoaponeurótico (Maf), ADN e outros compañeiros de transcrición, así como formar un complexo nuclear co encima UbcM2 que conxuga a ubiquitina [11], [12]. Neh2 contén dous motivos importantes coñecidos como DLG e ETGE, que son esenciais para a interacción entre Nrf2 e o seu regulador negativo Keap1 [13], [14].
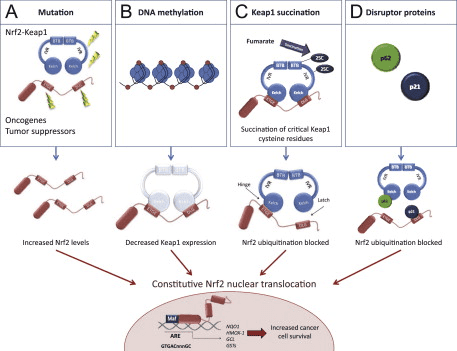
O Keap1 é un adaptador de sustrato para a ligase de Ubiquitina E3 baseado en cullin, que inhibe a actividade transcripcional de Nrf2 mediante ubiquitinación e degradación proteasómica en condicións normais [15], [16], [17]. Os dominios KELCH do homodímero Keap1 únense cos motivos DLG e ETGE do dominio Nrf2-Neh2 no citosol, onde ETGE actúa como bisagra con maior afinidade e DLG actúa como pestillo [18]. Baixo o estrés oxidativo ou despois da exposición aos activadores Nrf2, Nrf2 se disocia de Keap1 unión debido á modificación de tiol dos residuos de cisteína de Keap1 que ao final evitan a ubiquitinación de Nrf2 ea degradación proteasómica [19]. Entón Nrf2 se transloca cara ao núcleo, heterodimeriza con pequenas proteínas Maf e transacta unha batería ARE de xenes (Fig. 1A). O carboxi terminal de Neh3 actúa como dominio de transactivación interactuando co co-activador de transcrición coñecido como CHD6 (proteína de unión a ADN chromo-ATPase / helicase) [20]. Neh4 e Neh5 tamén actúan como dominios de transactivación, pero únense a outro co-activador transcripcional coñecido como CBP (cAMP-response-element binding proteína-binding protein) [21]. Ademais, Neh4 e Neh5 interactúan co cofactor nuclear RAC3 / AIB1 / SRC-3, levando a unha expresión xénica mellorada de Nrf2-targeted [22]. Neh5 ten un sinal de exportación nuclear sensible a redox que é crucial para a regulación e localización celular de Nrf2 [23].
Figura 1 Keap1 dependente e regulación independente de Nrf2. (A) En condicións basais, Nrf2 está secuestrado con Keap1 polos seus dous motivos (ETGE e DLG) que conduce á ubiquitinación mediada por CUL3 seguida da degradación do proteasoma. Baixo estrés oxidativo, Nrf2 disociase de Keap1, trasládase ao núcleo e activa a batería do xene ARE. (B) GSK3 fosforila Nrf2 e isto facilita o recoñecemento de Nrf2 por ?-TrCP para a ubiquitinación mediada por CUL1 e a posterior degradación do proteasoma. (C) p62 está secuestrado con Keap1, o que provoca a súa degradación autofáxica, a liberación de Nrf2 e o aumento da sinalización de Nrf2.
Regulación Nrf1 independente de Keap2
Evidencias emerxentes revelaron un novo mecanismo de regulación de Nrf2 que é independente de Keap1. O dominio Neh6 rico en serina de Nrf2 xoga un papel crucial nesta regulación ao unirse cos seus dous motivos (DSGIS e DSAPGS) á proteína que contén repetición de ?-transducina (?-TrCP) [24]. ?-TrCP é un receptor de substrato para o complexo de ubiquitina ligase Skp1�Cul1�Rbx1/Roc1 que se dirixe a Nrf2 para a ubiquitinación e a degradación do proteasoma. A glicóxeno sintase quinase-3 é unha proteína crucial implicada na estabilización e regulación de Nrf1 independente de Keap2; fosforila Nrf2 no dominio Neh6 para facilitar o recoñecemento de Nrf2 por ?-TrCP e a posterior degradación da proteína [25] (Fig. 1B).
Outros reguladores Nrf2
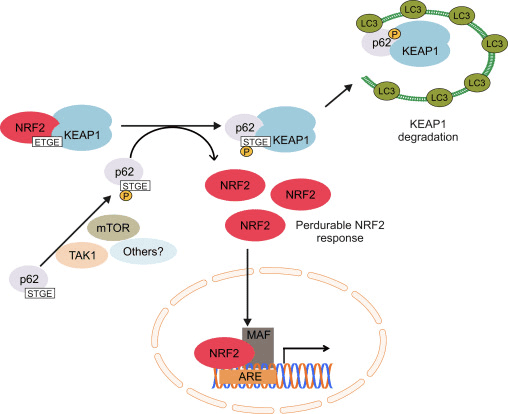
Outra liña de probas revelou unha vía non canónica de activación dependente de p62 Nrf2 na cal p62 secuestra Keap1 para a degradación autofágica que finalmente leva á estabilización de Nrf2 ea transactivación de xenes dependentes de Nrf2 [26], [27], [ 28], [29] (Fig. 1C).
A evidencia acumulada suxire que varios miARN xogan un papel importante na regulación da actividade de Nrf2 [30]. Sangokoya et al. [31] demostrou que o miR-144 regula directamente á baixa a actividade de Nrf2 na liña celular de linfoblastos K562, as células proxenitoras eritroides humanas primarias e os reticulocitos da enfermidade de células falciformes. Outro estudo interesante en células epiteliais de mama humana demostrou que miR-28 inhibe Nrf2 a través dun mecanismo independente de Keap1 [32]. Do mesmo xeito, os miRNAs como miR-153, miR-27a, miR-142-5p e miR144 regulan á baixa a expresión de Nrf2 na liña celular neuronal SH-SY5Y [33]. Singh et al. [34] demostraron que a expresión ectópica de miR-93 diminúe a expresión dos xenes regulados por Nrf2 nun modelo de carcinoxénese mamaria de rata inducida por 17 ?-estradiol (E2).
Un descubrimento recente do noso laboratorio identificou un inhibidor endóxeno de Nrf2 coñecido como receptor X retinoico alfa (RXR?). RXR? é un receptor nuclear, interactúa co dominio Neh7 de Nrf2 (residuos de aminoácidos 209-316) a través do seu dominio de unión ao ADN (DBD) e inhibe especificamente a actividade de Nrf2 no núcleo. Ademais, outros receptores nucleares, como o receptor activado polo proliferador de peroxisomas, o ER, o receptor relacionado con estróxenos e os receptores de glucocorticoides, tamén se informou de que son inhibidores endóxenos da actividade Nrf2 [9], [10].
Papel antiinflamatorio do eixe Nrf2 / HO-1
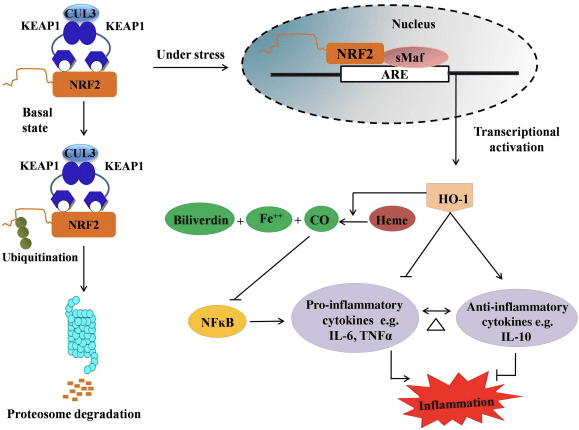
HO-1 é a isoforma inducible e unha enzima limitante de velocidade que cataliza a degradación do hemo en monóxido de carbono (CO) e ferro libre, e biliverdina a bilirrubina. A degradación enzimática do hemo libre proinflamatorio así como a produción de compostos antiinflamatorios como CO e bilirrubina desempeñan un papel importante no mantemento dos efectos protectores de HO-1 (Fig. 2).
Figura 2 Descrición xeral da ruta Nrf2 / HO-1. En condicións basais, Nrf2 únese ao seu represor Keap1 o que leva á ubiquitinación seguida da degradación do proteasoma. Durante o estrés oxidativo, o Nrf2 libre trasládase ao núcleo, onde se dimeriza con membros da pequena familia Maf e únese a xenes ARE como HO-1. O HO-1 regulado ao alza cataliza o hemo en CO, bilirrubina e ferro libre. O CO actúa como inhibidor da vía NF-?B que leva á diminución da expresión de citocinas proinflamatorias, mentres que a bilirrubina tamén actúa como antioxidante. Ademais, HO-1 inhibe directamente as citocinas proinflamatorias, así como activar as citocinas antiinflamatorias, polo que leva a equilibrar o proceso inflamatorio.
Nrf2 induce o xene HO-1 aumentando a expresión do ARNm e da proteína e é un dos xenes regulados Nrf2 clásicos que se usan amplamente en numerosos estudos in vitro e in vivo. Varios estudos demostraron que o HO-1 e os seus metabolitos teñen efectos antiinflamatorios significativos mediados por Nrf2. A elevación da expresión de HO-1 que está mediada polo Nrf2 activado leva á inhibición da sinalización de NF?B que produce unha redución da lesión da mucosa intestinal e unha disfunción da unión estreita no modelo de transplante de fígado de rata Sprague-Dawley macho [35]. A regulación positiva da expresión de HO-2 dependente de Nrf1 pode protexer os mioblastos C2C12 derivados do rato da citotoxicidade do H2O2 [36]. A HO-2 dependente de Nrf1 ten un impacto nas respostas inflamatorias mediadas por lipopolisacáridos (LPS) nos macrófagos de células espumosas derivadas de macrófagos peritoneais RAW264.7 ou de rato. A actividade Nrf2 desensibilizou o fenotipo dos macrófagos de células espumosas e prevén a inflamación desmedida dos macrófagos, estes desempeñan un papel importante na progresión da aterosclerose [37]. O eixe Nrf2/HO-1 afecta ás células microgliais BV2 do rato do rato e ás células HT22 do hipocampo do rato, con impacto na neuroinflamación. Regulación á alza da expresión de HO-1 a través da vía Nrf2 en células microgliais BV2 do rato que defenden a morte celular das células HT22 do hipocampo do rato [38]. Ademais, as moléculas híbridas a base de cobalto (HYCO) que combinan un inductor de Nrf2 cun liberador de monóxido de carbono (CO) aumentan a expresión de Nrf2/HO-1, liberan CO e exercen actividade antiinflamatoria in vitro. Os HYCO tamén regulan o tecido HO-1 e entregan CO no sangue despois da administración in vivo, apoiando o seu uso potencial contra condicións inflamatorias [39]. A regulación positiva de Nrf2/HO-1 reduce a inflamación ao aumentar a actividade eferocítica dos macrófagos murinos tratados con cloraminas de taurina [40]. En conxunto, os modelos experimentais explicados anteriormente revelaron que o eixe Nrf2/HO-1 xoga un papel importante na función antiinflamatoria, o que suxire que Nrf2 é un obxectivo terapéutico en enfermidades asociadas á inflamación.
Ademais, os subprodutos de HO-1 como CO, bilirrubina, actúan como un poderoso antioxidante durante o estrés oxidativo e danos celulares [41], [42]; suprime a encefalomielitis e a hepatite autoinmune [43], [44]; e protexe os ratos e os ratones contra o choque endotóxico evitando a xeración de iNOS e NO [45], [46], [47]. Por outra banda, a bilirrubina reduce a activación endotelial e a disfunción [48]. Curiosamente, a bilirrubina reduce a transmigración dos leucocitos endoteliais a través da molécula de adhesión 1 [49]. Estas referencias específicas que indican que HO-1 non só actúa como un potente axente antiinflamatorio, senón tamén os seus metabolitos.
Mediadores inflamatorios e encimas inhibidos por Nrf2
Citocinas e quimioquinas
As citocinas son proteínas e polipéptidos de baixo peso molecular secretados por unha variedade de células; regulan o crecemento celular, a diferenciación e a función inmune, e están implicados na inflamación e na cicatrización de feridas. As citocinas inclúen interleucinas (IL), interferóns, factor de necrose tumoral (TNF), factor estimulante de colonias, quimiocinas e factores de crecemento. Algunhas citocinas contan como mediadores proinflamatorios mentres que outras teñen funcións antiinflamatorias. A exposición ao estrés oxidativo produce unha sobreprodución de citocinas que provoca estrés oxidativo nas células diana. Varias citocinas proinflamatorias prodúcense en exceso cando o NF-?B é activado polo estrés oxidativo. Ademais, o estrés oxidativo proinflamatorio provoca unha maior activación do NF-?B e a sobreprodución de citocinas. A activación do sistema Nrf2/ARE xoga un papel importante na interrupción deste ciclo. As quimiocinas son unha familia de pequenas citocinas, cuxa función principal é guiar a migración das células inflamatorias. Funcionan principalmente como quimioatrayentes para leucocitos, monocitos, neutrófilos e outras células efectoras.
Informeuse de que a activación de Nrf2 impide a regulación transcripcional inducida por LPS das citocinas proinflamatorias, incluíndo IL-6 e IL-1? [50]. IL-1? e a produción de IL-6 tamén se incrementa en Nrf2?/? ratos con colite inducida por sulfato de dextrano [51], [52]. Nrf2 inhibe a produción de IL-17 augas abaixo e outros factores inflamatorios Th1 e Th17, e suprime o proceso da enfermidade nun modelo experimental de esclerose múltiple, encefalite autoinmune [53]. Os xenes antioxidantes dependentes de Nrf2 HO-1, NQO-1, Gclc e Gclm bloquean TNF-?, IL-6, a proteína quimio-atractora de monocitos-1 (MCP1), a proteína inflamatoria de macrófagos-2 (MIP2) e a proteína inflamatoria. mediadores. Pero no caso dos ratos knockout Nrf2, o efecto antiinflamatorio non se produce [54]. Os neutrófilos peritoneais de ratos knockout Nrf2 tratados con LPS teñen niveis significativamente máis altos de citocinas (TNF-a e IL-6) e quimiocinas (MCP1 e MIP2) que as células de tipo salvaxe (WT) [54]. In vitro, a transferencia do xene Nrf2 ás células do músculo liso da aorta humana e do coello suprime a secreción de MCP1 [8], [55], e a expresión de HO-2 dependente de Nrf1 suprime NF-?B e MCP-1 estimulados por TNF-? secreción nas células endoteliais da vea umbilical humana [56]. Estes achados apuntan a que, en resposta a estímulos inflamatorios, a regulación positiva da sinalización de Nrf2 inhibe a sobreprodución de citocinas e quimiocinas proinflamatorias, así como limita a activación de NF-?B.
Moléculas de adhesión celular
As moléculas de adhesión celular (CAM) son proteínas que se unen ás células ou á matriz extracelular. Situados na superficie celular, están implicados no recoñecemento celular, a activación celular, a transdución de sinais, a proliferación e a diferenciación. Entre as CAM, ICAM-1 e VCAM-1 son membros importantes da superfamilia das inmunoglobulinas. O ICAM-1 está presente en baixas concentracións nos leucocitos e nas membranas das células endoteliais. Tras a estimulación das citocinas, a concentración aumenta significativamente. O ICAM-1 pode ser inducido pola IL-1 e o TNF e exprésase polo endotelio vascular, os macrófagos e os linfocitos. É un ligando para a integrina, un receptor que se atopa nos leucocitos. Cando se activa a ponte ICAM-1-integrina, os leucocitos únense ás células endoteliais e despois migran aos tecidos subendoteliais [57]. VCAM-1 media na adhesión de linfocitos, monocitos, eosinófilos e basófilos ao endotelio vascular e contribúe ao recrutamento de leucocitos, o que finalmente conduce a danos nos tecidos debido ao estrés oxidativo. Nrf2 inhibe a actividade promotora de VCAM-1 [58]. O xene HO-2 regulado por Nrf1 pode afectar a expresión de E-selectina e VCAM-1, moléculas de adhesión asociadas ás células endoteliais [59]. A expresión pulmonar de varias CAM como CD-14, TREM1, SELE, SELP e VCAM-1 é significativamente maior en Nrf2?/? ratos que en ratos Nrf2+/+ [60]. Nrf2 nas células endoteliais da aorta humana suprime a expresión de VCAM-1 inducida por TNF-β e interfire coa adhesión das células monocíticas U937 inducida por TNF-β [8]. A sobreexpresión de Nrf2 tamén inhibe a expresión do xene VCAM-1 inducida por TNF-β nas células endoteliais microvasculares humanas [61]. O ácido 3-hidroxiantranilico (HA), un antioxidante natural, un dos metabolitos de l-triptófano formado in vivo ao longo da ruta metabólica coñecida como vía da quinurenina durante a inflamación ou a infección, induce a expresión de HO-1 e estimula a Nrf2 no umbilical humano. células endoteliais venosas (HUVEC). A expresión de HO-2 dependente de Nrf1 inducida polo HA inhibe a secreción de MCP-1, a expresión de VCAM-1 e a activación de NF-kB asociada á lesión vascular e á inflamación na aterosclerose [56]. A calcona sintética antiproliferativa e antiinflamatoria derivada 2?,4?,6?-tris (metoximetoxi) calcona inhibe ICAM-1, a citocina proinflamatoria IL-1? e TNF-? expresión no tecido colónico de ratos tratados con ácido trinitrobenceno sulfónico [62]. A regulación positiva de Nrf2 inhibe a expresión de ICAM-1 inducida por TNF-β nas células epiteliais pigmentarias da retina humana tratadas con licopeno [63]. Todos estes estudos suxiren que Nrf2 xoga un papel fundamental no proceso inflamatorio ao regular a migración e infiltración das células inflamatorias ao tecido inflamado.
Matrix Metalloproteinases (MMPs)
As MMP están moi presentes na matriz extracelular e están implicadas en procesos fisiolóxicos e patolóxicos como a proliferación celular, migración, diferenciación, cicatrización de feridas, anxioxénese, apoptose e metástase tumoral. Informeuse de que o eixe Nrf2/HO-1 inhibe a MMP-9 nos macrófagos e a MMP-7 nas células epiteliais intestinais humanas, e isto é beneficioso no tratamento da enfermidade inflamatoria intestinal [62], [64]. O dano cutáneo inducido pola irradiación UV é máis grave nos ratos con Nrf2-knockout que nos ratos WT e o nivel de MMP-9 é significativamente maior, o que indica que Nrf2 reduce a expresión de MMP-9. Polo tanto, considérase que Nrf2 é protector contra a irradiación UV [65]. Outro estudo tamén informou de que a activación transcripcional regulada á baixa de MMP-9 na invasión e inflamación das células tumorais está regulada mediante a inhibición da vía de sinalización NF-kB [66]. Na lesión traumática da medula espiñal, a vía de sinalización NF-kB tamén participa na regulación dos niveis de ARNm de MMP-9 [67]. Polo tanto, na inflamación a regulación das MMP vese afectada directamente pola vía Nrf2 ou indirectamente pola vía NF-?B influenciada por Nrf2.
Ciclooxigenasa-2 (COX2) e óxido nítrico inducible sintase (INOS)
Unha serie de experimentos en ratos knockout Nrf2 demostraron o seu papel crucial na inflamación e na regulación de xenes proinflamatorios como COX-2 e iNOS. Por primeira vez, Khor et al. informou de aumento da expresión de citocinas proinflamatorias como COX-2 e iNOS nos tecidos colónicos de Nrf2?/? ratos en comparación cos ratos WT Nrf2+/+, o que indica que Nrf2 suprime a súa actividade [51]. Outro informe sobre o pretratamento con sulforafano, un dos coñecidos activadores de Nrf2 presentes en vexetais crucíferas, demostrou o seu efecto antiinflamatorio de inhibir a expresión de TNF-?, IL-1?, COX-2 e iNOS tanto no ARNm. e niveis de proteína en macrófagos peritoneais primarios de ratos Nrf2+/+ en comparación cos de Nrf2?/? ratos [68]. Do mesmo xeito, o hipocampo dos ratos knockout Nrf2 con inflamación inducida por LPS tamén mostra unha maior expresión de marcadores de inflamación como iNOS, IL-6 e TNF-? que os ratos WT [69]. Así mesmo, os ratos knockout Nrf2 son hipersensibles ao estrés oxidativo inducido pola 1-metil-4-fenil-1,2,3,6-tetrahidropiridina, ademais de mostrar un aumento dos niveis de ARNm e proteínas de marcadores de inflamación como COX-2, iNOS. , IL-6 e TNF-? [70]. Ademais, os fígados de Nrf2?/? Os ratos sometidos a unha dieta deficiente en metionina e colina teñen unha expresión de ARNm de Cox5 e iNOS 2 veces máis alta que os dos ratos WT coa mesma dieta, o que suxire un papel antiinflamatorio de Nrf2 [71]. Recentemente, Kim et al. demostraron que o etilpiruvato fitoquímico exerce os seus efectos antiinflamatorios e antioxidantes ao diminuír a expresión de iNOS mediante a sinalización Nrf2 nas células BV2. Demostraron que o etil piruvato induce a translocación nuclear de Nrf2, que finalmente inhibe a interacción entre p65 e p300, o que leva a unha diminución da expresión de iNOS [72]. Ademais, o análogo de carbazol LCY-2-CHO activa a Nrf2 e provoca a súa translocación nuclear, o que leva á supresión da expresión de COX2 e iNOS [73] nas células musculares lisas vasculares da aorta de rata.
Papel paradoxal de Nrf2 na regulación da actividade NLRP3 iIflammasoma
A familia NLR, o dominio de pirina que contén 3 (NLRP3) inflamasoma é un complexo multiproteico que funciona como un receptor de recoñecemento de patóxenos (PRR) e recoñece a gran variedade de sinais microbianos de estrés oxidativo, como os patróns moleculares asociados a patóxenos (PAMP), os danos. moléculas de patrón molecular asociadas (DAMP) e ROS [74]. O inflamasoma NLRP3 activado media a escisión da caspase-1 e a secreción da citocina proinflamatoria interleucina-1? (IL-1?) que finalmente induce o proceso de morte celular coñecido como piroptose que protexe aos hóspedes contra unha ampla gama de patóxenos [75]. Non obstante, a activación aberrante do inflamasoma está asociada con enfermidades de plegamento incorrecto das proteínas, como encefalopatías esponxiformes transmisibles, enfermidade de Alzheimer, enfermidade de Parkinson e tamén diabetes tipo 2 [76], cancro [77], gota e aterosclerose [78].
Unha observación recente do grupo Rong Hu sobre a asociación de Nrf2 coa regulación negativa do inflamasoma revelou que, Nrf2 induce a expresión de NQO1 que leva á inhibición da activación do inflamasoma NLRP3, a escisión da caspase-1 e a IL-1? xeración en macrófagos. Ademais, un activador de Nrf2 ben coñecido, a terc-butilhidroquinona (tBHQ) regulaba negativamente a transcrición de NLRP3 activando o ARE de forma dependente de Nrf2 [79]. Ademais da observación anterior, o mesmo grupo tamén se revelou que o fumarato de dimetilo (DMF) prevén a colite inducida por DSS mediante a activación da vía de sinalización Nrf2 que está implicada na translocación nuclear de Nrf2 e na inhibición do ensamblaxe do inflamasoma NLRP3 [80].
Unha serie de experimentos con compostos naturais e sintéticos tamén revelaron o efecto inhibidor de Nrf2 na activación do inflamasoma NLRP3. Por exemplo, o tratamento da epigalocatequina-3-galato (EGCG) en ratos con nefrite lúpica mostrou unha diminución da activación do inflamasoma renal NLRP3, que está mediada pola vía de sinalización Nrf2 [81]. Así mesmo, o citral (3,7-dimetil-2,6-octadienal), un dos principais compostos activos dunha herba medicinal chinesa Litsea cubeba, inhibe a activación do inflamasoma NLRP3 a través da vía de sinalización antioxidante Nrf2 no modelo de rato de nefrite lúpica acelerada e severa (ASLN). [82]. Do mesmo xeito, a biocanina protexeu contra a lesión hepática inducida por LPS/GalN activando a vía Nrf2 e inhibindo a activación do inflamasoma NLRP3 en ratos machos BALB/c [83]. Ademais, tamén se demostrou que a mangiferina regula a expresión de Nrf2 e HO-1 de forma dependente da dose e inhibe NLRP3 hepático inducido por LPS/D-GalN, ASC, caspase-1, IL-1? e TNF-? expresión [84].
A pesar da regulación negativa de NLRP3 por Nrf2, tamén activa a función inflamasoma NLRP3 e AIM2. Haitao Wen e os seus colegas descubriron que, Nrf2 ?/? Os macrófagos de rato mostraron a activación defectuosa do inflamasoma NLRP3 e AIM2 pero non do inflamasoma NLRC4 [85]. Curiosamente, esta observación está a representar as funcións descoñecidas de Nrf2 no contexto das enfermidades asociadas á inflamación; por iso é moi importante estudar máis para revelar o mecanismo no que Nrf2 activa a función inflamasoma antes de considerala como unha diana terapéutica.
Supresión da transcripción de citocinas pro-inflamatorias por Nrf2
Unha investigación moi recente baseada nos resultados de inmunoprecipitación da cromatina (ChIP)-seq e ChIP-qPCR en macrófagos de rato revelou que Nrf2 únese ás rexións promotoras de citocinas proinflamatorias como a IL-6 e a IL-1? e inhibe o recrutamento de ARN Pol II. Como resultado, o ARN Pol II non é capaz de procesar a activación transcripcional de IL-6 e IL-1? que finalmente leva á inhibición da expresión xénica. Por primeira vez, o grupo de Masayuki Yamamoto revelou o novo mecanismo polo cal Nrf2 non só transactiva os seus xenes augas abaixo a través de ARE, senón que tamén suprime a activación transcripcional de xenes específicos con ou sen ARE inhibindo o recrutamento de ARN Pol II [50].
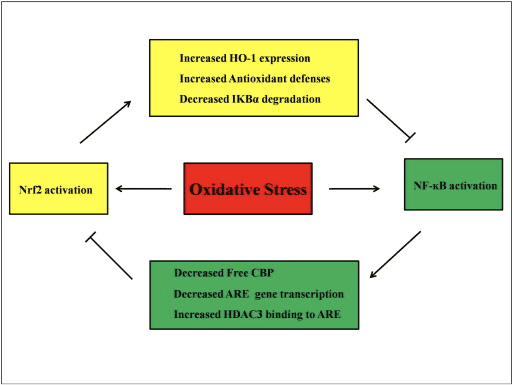
Diafonía entre as vías Nrf2 e NF-?B
NF-?B é un complexo proteico responsable da transcrición do ADN que se atopa en case todos os tipos de células animais e está implicado en diversos procesos como a inflamación, a apoptose, a resposta inmune, o crecemento celular e o desenvolvemento. A p65, unha proteína Rel da familia NF-?B, ten un dominio de transactivación mentres que a p50 non o ten e require heterodimerización coa proteína Rel para activar a transcrición. Durante o estrés oxidativo, a I?B quinase (IKK) actívase e provoca a fosforilación de I?B, dando como resultado a liberación e a translocación nuclear de NF-?B. NF-?B provoca a transcrición de mediadores proinflamatorios como IL-6, TNF-?, iNOS, IL-1 e a adhesión intracelular COX-2.
A regulación anormal de NF-?B relacionouse coa artrite reumatoide, o asma, a enfermidade inflamatoria intestinal e a gastrite inducida pola infección por Helicobacter pylori [86]. Actualmente considérase que a actividade de NF-kB inflúe na vía de sinalización Keapl/Nrf2/ARE principalmente en tres aspectos: primeiro, Keap1 degrada IKK? a través da ubiquitinación, inhibindo así a actividade de NF-?B [87]. En segundo lugar, o proceso inflamatorio induce mediadores inflamatorios como a COX2 derivada da ciclopentenona prostaglandina 15d-PGJ2, un forte electrófilo que reacciona con Keap1 e activa Nrf2, iniciando así a transcrición xenética coa inhibición simultánea da actividade de NF-kB [58], [88] ( Fig. 3 A, B). En terceiro lugar, NF-?B pode combinarse co co-activador transcripcional competitivo Nrf2 CBP [89], [90] (Fig. 3 C, D).
Figura 3 Diafonía entre as vías Nrf2 e NF-?B. (A) Keap1 dirixe o IKK á ubiquitinación mediada por CUL3 e á degradación do proteasoma que finalmente conduce á inhibición da fosforilación de NF-?B e este mecanismo tamén funciona como unión competitiva de Nrf2 e IKK con Keap1. (B) O estrés oxidativo activa a IKK que fosforila NF-?B, o que leva á súa translocación ao núcleo e á activación de citocinas proinflamatorias como a COX-2. O produto terminal de COX-2 coñecido como 15d-PGJ2 actúa como un inductor de Nrf2 que finalmente conduce á supresión do estrés oxidativo. (C) Nrf2 únese co seu cofactor de transcrición CBP xunto con pequenos Maf e outra maquinaria de transcrición para iniciar a expresión xenética dirixida por ARE. (D) Cando NF-?B únese á CBP de forma competitiva, inhibe a unión da CBP a Nrf2, o que leva á inhibición da transactivación de Nrf2.
Suponse que as vías de sinalización Nrf2 e NF-?B interactúan para controlar a transcrición ou a función das proteínas diana augas abaixo. Para xustificar esta suposición, moitos exemplos mostran que a activación e inhibición directa ou indirecta ocorre entre os membros das vías Nrf2 e NF-?B (Fig. 4). En resposta ao LPS, o knockdown de Nrf2 aumenta significativamente a actividade transcripcional de NF-?B e a transcrición do xene dependente de NF-?B, mostrando que Nrf2 impide a actividade de NF-?B [60], [91]. Ademais, o aumento da expresión de HO-2 augas abaixo dependente de Nrf1 inhibe a actividade de NF-?B. Cando as células do cancro de próstata se exponen brevemente ao succinato de ?-tocoferilo, un derivado da vitamina E, a expresión de HO-1 está regulada. Os produtos finais de HO-1 inhiben a translocación nuclear de NF-?B [92]. Estes estudos in vivo suxiren que Nrf2 regula negativamente a vía de sinalización NF-kB. O LPS estimula a actividade de unión ao ADN de NF-?B e o nivel da subunidade p65 de NF-?B é significativamente maior nos extractos nucleares dos pulmóns de Nrf2?/? que dos ratos WT, o que suxire un papel negativo de Nrf2 na activación de NF-?B. Ademais, Nrf2?/? fibroblastos de embrións de rato tratados con LPS e TNF-? mostran unha activación de NF-?B máis destacada causada pola activación de IKK e de I?B-? degradación [60]. E a eliminación do virus respiratorio sincitial diminúe significativamente mentres que a actividade de unión ao ADN de NF-?B aumenta en Nrf2?/? ratos en comparación cos ratos WT [93]. Nefrite lúpica inducida por pristano en Nrf2?/? Os ratos cotratados con sulforafano teñen danos renales graves e alteracións patolóxicas, así como unha expresión elevada de iNOS e activación de NF-?B en comparación co WT, o que suxire que Nrf2 mellora a nefrite lúpica inhibindo a vía de sinalización de NF-?B e eliminando ROS [94]. ]. A actividade de NF-?B tamén ocorre cando as células son tratadas cun inductor de Nrf2 xunto con LPS e TNF-?. Por exemplo, un derivado sintético de calcona inhibe a activación de NF-?B inducida por TNF-β tanto directa como indirectamente e en parte mediante a indución da expresión de HO-1 nas células HT-29 do epitelio intestinal humano [62]. A supresión da translocación de NF-?B e da actividade de unión ao ADN, así como a supresión da expresión de iNOS nos hepatocitos, atópanse cando as ratas F344 son tratadas con 3H-1,2-ditiol-3-tiona (D3T) [95]. Despois do cotratamento con sulforafano e LPS, a expresión inducida por LPS de iNOS, COX-2 e TNF-? en Raw 264.7 macrófagos está regulado á baixa, o que suxire que o sulforafano ten actividade antiinflamatoria mediante a inhibición da unión ao ADN de NF-?B [96]. Aínda que se realizaron varios estudos experimentais para explicar o vínculo entre as vías Nrf2 e NF-?B, aínda quedan resultados conflitivos. Tanto as normativas positivas como negativas foron reportadas entre Nrf2 e NF-kB [97]. Normalmente, os electrófilos quimiopreventivos 3H-1,2-ditiol-3-tiona o sulforafano e triterpenóides CDDO-Me activar Nrf2 través da inhibición de NF-kB e os seus xenes regulados abaixo [98] [99] [100]. En contraste, varios axentes ou condicións como o ROS, o LPS, o estrés de cisallamento de fluxo, o LDL oxidado e o fume de cigarro demostraron aumentar a actividade de Nrf2 e NF-kB [97]. Ademais, estudos in vivo revelaron que a actividade de NF-kB diminúe nos fígados illados de Nrf2?/? ratos e actividade de unión NF-?B é menor en Nrf2?/? que nos ratos Nrf2+/+ [101]. Non obstante, as células endoteliais aórticas humanas tratadas co vector adenoviral Nrf2 inhiben os xenes de NF-?B abaixo sen afectar a actividade de NF-?B [8].
Figura 4 Bucle regulador de Nrf2 e NF-?B. A vía Nrf2 inhibe a activación de NF-?B evitando a degradación de I?B-? e aumentar a expresión de HO-1 e as defensas antioxidantes que neutralizan ROS e produtos químicos desintoxicantes. Como resultado, suprime a activación de NF-?B asociada a ROS. Así mesmo, a transcrición mediada por NF-?B reduce a activación de Nrf2 reducindo�SANA transcrición xenética e a proteína libre de unión a CREB competindo con Nrf2 pola CBP. Ademais, NF-?B aumenta o recrutamento de histona desacetilasa (HDAC3) para a rexión ARE e, polo tanto, impide a activación transcripcional de Nrf2.
A activación da vía de sinalización Nrf2 desempeña un papel importante na expresión de encimas e xenes implicados na desintoxicación dos oxidantes reactivos aumentando a capacidade antioxidante das células no corpo humano. Aínda que hai moitos estudos de investigación dispoñibles hoxe, os mecanismos reguladores da activación Nrf2 non se entenden completamente. Tamén se atopou un posible papel da vía de sinalización Nrf2 no tratamento da inflamación. Dr. Alex Jimenez DC, CCST Insight
Papel da Nrf2 en enfermidades inflamatorias
Os estudos in vivo demostraron que Nrf2 desempeña un papel importante nas enfermidades inflamatorias que afectan a diferentes sistemas; Estes inclúen gastrite, colite, artrite, neumonía, dano ao fígado, enfermidades cardiovasculares, enfermidades neurodegenerativas e danos cerebrais. Nestes estudos, Nrf2?/? os animais mostraron síntomas máis graves de inflamación e danos nos tecidos que os animais WT. Polo tanto, crese que a vía de sinalización Nrf2 ten un efecto protector nas enfermidades inflamatorias. A instalación intra-traqueal da elastase pancreática porcina induce a enfermidade pulmonar obstrutiva crónica, especialmente o enfisema. Os ratos deficientes con Nrf2 son altamente susceptibles ao enfisema e a redución da expresión de HO-1, PrxI eo xene de antiproteasa SLPI ocorre en macrófagos alveolares. Nrf2 é considerado un regulador clave no sistema de defensa mediada por macrófagos contra a lesión pulmonar [102]. Ratinho Nrf2 deficiente con enfisema inducido por exposición ao fume de tabaco por mes 6 mostran aumento da inflamación broncoalveolar, tarada positivamente a expresión de marcadores de estrés oxidativo en alvéolos, e aumento da apoptose de células do septo alveolar, o que suxire que Nrf2 actúa contra o enfisema inducido por fume a través do aumento da expresión de antioxidante xenes [102], [103]. Con Nrf2 interrupción, inflamación das vías aéreas mediadas por alérgeno e asma utilizando complexo de inflamación ovalbumina mostra aumentada das vías aéreas, hiper-reactividade, hiperplasia das células caliciformes, e altos niveis de Th2 no lavado broncoalveolar e esplenócitos, mentres que os límites da vía de sinalización Nrf2 mediadas por vías aéreas eosinofilia , hipersecreción mucosa e hiperreactividade das vías respiratorias, así como inducir moitos xenes antioxidantes que impiden o desenvolvemento do asma [104]. A inxección de carragenina na cavidade pleural induce a pleurisy, ea acumulación 15d-PGJ2 nas células inflamatorias Nrf2 está limitada aos macrófagos peritoneales do rato. Durante a fase inicial da inflamación, 15d-PGJ2 activa Nrf2 e regula o proceso inflamatorio mediante a indución de HO-1 e PrxI. Un estudo tamén suxeriu que COX-2 ten un efecto antiinflamatorio na fase inicial pola produción de 15d-PGJ2 [105]. A administración oral de xNUMX% de dextrano sulfato de sodio durante 1 semana induce a colite asociada a alteracións histolóxicas que inclúen acortadores de criptas e infiltración de células inflamatorias no tecido do colon. Para protexer a integridade intestinal na colite, Nrf2 pode desempeñar un papel importante regulando citoquinas proinflamatorias e indución de enzimas desintoxicantes na fase II [51]. Nun modelo de rato Nrf2-knockout de sepsis pulmonar inducida por LPS, a actividade de NF-?B regula a influencia de citocinas inflamatorias como COX-2, IL-113, IL-6 e TNF? que son esenciais para iniciar e promover a inflamación [60]. Nrf2 reduce o dano inflamatorio regulando estes factores inflamatorios. Nestes modelos de inflamación aguda, a maior regulación das enzimas antioxidantes, as citoquinas proinflamatorias e os mediadores pola vía de sinalización Nrf2 reduce a lesión inflamatoria nos animais WT. Curiosamente, isto tamén se informou nos ratones Nrf2-knockout nos que os síntomas son marcadamente exacerbados en comparación cos ratones WT.
Investigación sobre fármacos antiinflamatorios dependentes de Nrf2
En resumo, comentamos experimentos que demostran que a vía de sinal Nrf2 desempeña un papel regulador en moitas áreas de inflamación, polo tanto, os axentes antiinflamatorios dependentes de Nrf2 son importantes para o tratamento de enfermidades inflamatorias.
As plantas foron fontes extraordinariamente ricas de compostos que activan o factor de transcrición Nrf2, que conduce á regulación reguladora dos xenes citoprotectores. Recentemente, realizáronse varios estudos para investigar os efectos de diferentes axentes antiinflamatorios, principalmente de orixe vexetal. Por exemplo, a curcumina é o ingrediente activo da azafrán e tamén se atopa en pequenas cantidades en xenxibre; Os isotiocianatos, especialmente os fenilisotiocianatos, son do brócoli, o apio e outros vexetais; e as antocianinas son de froitas e uvas [124]. Os estudos demostraron que todos estes axentes non só son bos antioxidantes, pero tamén teñen efectos antiinflamatorios potentes a través da indución Nrf2 [125], [126]. Polo tanto, o desenvolvemento de novos activadores antiinflamatorios de Nrf2 a partir de extractos de plantas atraeu moito interese na investigación médica.
Nos últimos anos realizáronse moitos experimentos con animais para confirmar as accións destes compostos. O artesunato úsase principalmente para a malaria grave, a malaria cerebral e as enfermidades autoinmunes reumáticas; tamén é eficaz na lesión pulmonar séptica. O artesunato activa a expresión de Nrf2 e HO-1, e esta última reduce a entrada de citocinas e leucocitos proinflamatorios ao tecido para previr a inflamación [127]. Pénsase que a isovitexina, extraída das cascas do arroz Oryza sativa, ten propiedades antiinflamatorias e antioxidantes; xoga un papel protector contra a lesión pulmonar aguda inducida polo LPS activando a vía Nrf2/HO-1 e inhibindo a MAPK e NF-?B [128]. O fimasartan, un bloqueador do receptor de angiotensina II recentemente popular que actúa sobre o sistema renina-anxiotensina, reduce a presión arterial; o uso de fimasartan para tratar ratos con obstrución ureteral unilateral inducida cirurxicamente reduce o estrés oxidativo, a inflamación e a fibrose mediante a regulación positiva de Nrf2 e a vía antioxidante e inhibindo RAS e MAPK [129]. A sapanona distribúese amplamente no sueste asiático, onde se usa como medicamento antigripal, antialérxico e neuroprotector; activa Nrf2 e inhibe NF-?B polo que pode ser beneficioso no tratamento de enfermidades relacionadas con Nrf2 e/ou NF-?B [130]. A bixina extraída das sementes de Bixin orellana úsase para enfermidades infecciosas e inflamatorias en México e América do Sur; diminúe os mediadores inflamatorios, a fuga dos capilares alveolares e o dano oxidativo de forma dependente de Nrf2 para aliviar a lesión pulmonar inducida pola ventilación e restaurar a morfoloxía pulmonar normal [131]. Outros compostos vexetais, como o galato de epigalocatequina, o sulforafano, o resveratrol, o licopeno e o extracto de té verde teñen efectos terapéuticos sobre enfermidades inflamatorias a través da vía de sinalización Nrf2 [132], [133], [134]. Recentemente, outro fitoquímico, o eriodictiol, que está presente nos cítricos, ten efectos antiinflamatorios e antioxidantes sobre a lesión renal inducida por cisplatino e a lesión pulmonar aguda inducida pola sepsis ao regular Nrf2, inhibindo NF-?B e inhibindo o expresión de citocinas en macrófagos [135], [136]. Non obstante, numerosos fitoquímicos mostran unha gran promesa para a prevención e o tratamento de diversas enfermidades humanas, e algúns xa entraron na fase de ensaios clínicos (táboa 2).
Estes compostos vexetais activan a vía de sinalización Nrf2 principalmente en forma de materiais electrofílicos que modifican os residuos de cisteína de Keap1, levando a un enlace nuclear Nrf2 libre co ARE, obtendo a activación da transcrición do xene correspondente.
Sulforaphane e os seus efectos sobre o cancro, a mortalidade, o envellecemento, o cerebro e o comportamento, as enfermidades cardíacas e moito máis
Os isotiocianatos son algúns dos compostos vexetais máis importantes que pode obter na súa dieta. Neste video fago o caso máis completo para eles que se fixo. ¿A atención curta? Saltar ao teu tema favorito premendo un dos puntos de tempo a continuación. Cadro de cronograma completo a continuación.
Seccións clave:
00: 01: 14 - Cáncer e mortalidade
00: 19: 04 - Envellecemento
00: 26: 30 - Cerebro e comportamento
00: 38: 06 - Recapitalización final
00: 40: 27 - Dose
Cadro de tempo completo:
00: 00: 34 - Introdución de sulforaphane, un foco principal do vídeo.
00: 01: 14 - Consumo e redución de vexetais cruciferos na mortalidade por todas as causas.
00: 02: 12 - Risco de cancro de próstata.
00: 02: 23 - Risco de cancro de vejiga.
00: 02: 34 - Cáncer de pulmón en risco de fumadores.
00: 02: 48 - Risco de cancro de mama.
00: 03: 13 - hipotético: e se xa ten cancro? (intervencionista)
00: 03: 35 - Mecanismo plausible que conduce os datos asociativos de cancro e mortalidade.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Evidencia animal que mostra un forte efecto do extracto de brotes de brócoli no desenvolvemento de tumores vesicales en ratas.
00: 06: 06 - Efecto da suplementación directa de sulforaphane en pacientes con cancro de próstata.
00: 07: 09 - Bioacumulación de metabolitos de isotiocianato no tecido de mama real.
00: 08: 32 - Inhibición das células nais do cancro de mama.
00: 08: 53 - Lección de historia: as brassicas establecéronse con propiedades de saúde mesmo na Roma antiga.
00: 09: 16 - A capacidade de Sulforaphane para mellorar a excreción de carcinóxenos (benceno, acroleína).
00: 09: 51 - NRF2 como un cambio xenético a través de elementos de resposta antioxidante.
00: 10: 10 - Como a activación de NRF2 aumenta a excreción de carcinóxenos a través de glutatión-S-conjugados.
00: 10: 34 - As coles de Bruxelas aumentan a glutatión-S-transferasa e reducen o dano do ADN.
00: 11: 20 - A bebida de brote de brócoli aumenta a excreción de benceno por 61%.
00: 13: 31 - O homogeneio de brotes de brócoli aumenta as encimas antioxidantes nas vías aéreas superiores.
00: 15: 45 - Consumo de vexetais cruciferos e mortalidade cardíaca.
00: 16: 55 - O polbo de brócolis mellora os lípidos sanguíneos eo risco de enfermidade cardíaca en diabéticos tipo 2.
00: 19: 04 - Comezo da sección de envellecemento.
00: 19: 21 - A dieta enriquecida con Sulforaphane mellora a vida útil dos escaravellos de 15 a 30% (en certas condicións).
00: 20: 34 - Importancia da baixa inflamación por lonxevidade.
00: 22: 05 - As verduras cruciferas e os brotes de brócoli parecen reducir unha gran variedade de marcadores inflamatorios en humanos.
00: 23: 40 - Recapitalización media: cancro, seccións de envellecemento
00: 24: 14 - Os estudos do rato suxiren que o sulforaphano pode mellorar a función inmune adaptativa na vellez.
00: 25: 18 - Sulforaphane mellorou o crecemento do cabelo nun modelo de calvície de rato. Imaxe en 00: 26: 10.
00: 26: 30 - Comezo da sección do cerebro e do comportamento.
00: 27: 18 - Efecto do extracto de brotes de brócoli no autismo.
00: 27: 48 - Efecto da glucoraphanina na esquizofrenia.
00: 28: 17 - Inicio da discusión de depresión (mecanismo plausible e estudos).
00: 31: 21 - O estudo do rato usando 10 diferentes modelos de depresión inducida polo estrés mostran sulforaphane igualmente efectivo como a fluoxetina (prozac).
00: 32: 00 - O estudo mostra que a inxestión directa de glucoraphanina en ratos é igualmente eficaz na prevención da depresión do modelo de estrés da derrota social.
00: 33: 01 - Inicio da sección de neurodegeneración.
00: 33: 30 - Sulforaphane e enfermidade de Alzheimer.
00: 33: 44 - Sulforaphane e enfermidade de Parkinson.
00: 33: 51 - Sulforaphane e enfermidade de Hungtington.
00: 34: 13 - Sulforaphane aumenta as proteínas de choque térmico.
00: 34: 43 - Inicio da sección traumática de lesións cerebrais.
00: 35: 01 - Sulforaphane inxectado inmediatamente despois de que o TBI mellore a memoria (estudo do rato).
00: 35: 55 - Sulforaphane e plasticidade neuronal.
00: 36: 32 - Sulforaphane mellora a aprendizaxe en modelo de diabetes tipo II en ratos.
00: 37: 19 - Distrofia muscular sulforaphana e duxena.
00: 37: 44 - Inhibición da myostatina nas células satélite do músculo (in vitro).
00: 38: 06 - Recapitulación de última hora: mortalidade e cancro, danos no ADN, estrés oxidativo e inflamación, excreción de benceno, enfermidade cardiovascular, diabetes tipo II, efectos sobre o cerebro (depresión, autismo, esquizofrenia e neurodegeneración), vía NRF2.
00: 40: 27 - Pensamentos en descubrir unha dose de brotes de brócoli ou sulforaphane.
00: 41: 01 - Anécdotas sobre o xermelo na casa.
00: 43: 14 - Sobre as temperaturas de cocción e actividade sulforaphane.
00: 43: 45 - Conversión de bacterias gut de sulforaphane a partir de glucoraphanin.
00: 44: 24 - Os suplementos funcionan mellor cando se combinan con mirosinasa activa de vexetais.
00: 44: 56 - Técnicas de cociña e vexetais crucíferos.
00: 46: 06 - Isotiocianatos como goitrógenos.
Conclusións
Actualmente, moitas investigacións centráronse no papel da vía de sinalización Nrf2/Keap1/ARE na inflamación. Entre os encimas regulados pola Nrf2, HO-1 é un dos encimas representativos de resposta ao estrés. HO-1 ten propiedades antiinflamatorias e antioxidantes destacadas. En xeral, a vía de sinalización Nrf2 tamén regula negativamente as citocinas, os factores liberadores de quimiocinas, as MMPs e outros mediadores inflamatorios COX-2 e a produción de iNOS, que afectan directa ou indirectamente ás vías relevantes de NF-kB e MAPK e outras redes que controlan a inflamación. Suxírese que as vías de sinalización Nrf2 e NF-?B interactúan para regular a transcrición ou a función das proteínas diana augas abaixo. A supresión ou inactivación da actividade transcripcional mediada por NF-?B a través de Nrf2 probablemente ocorre na fase inicial da inflamación, xa que NF-?B regula a síntese de novo dunha serie de mediadores proinflamatorios. Non obstante, aínda hai algunhas limitacións na investigación, como se hai conexións entre Nrf2 e outras vías de sinalización como JAK/STAT, a importancia dos actuais activadores de Nrf2 derivados de fontes vexetais naturais na inflamación e como mellorar a actividade biolóxica. e mellorar a orientación destes compostos. Estes requiren unha validación experimental adicional.
Ademais, a vía de sinalización Nrf2 pode regular > 600 xenes [163], dos cales > 200 codifican proteínas citoprotectoras [164] que tamén están asociadas con inflamación, cancro, enfermidades neurodexenerativas e outras enfermidades importantes [165]. Crecentes evidencias suxiren que a vía de sinalización de Nrf2 está desregulada en moitos cancros, o que dá lugar a unha expresión aberrante da batería do xene Nrf2 dependente. Ademais, a inflamación xoga un papel importante nas enfermidades relacionadas co estrés oxidativo, especialmente no cancro. A aplicación de varios activadores de Nrf2 para contrarrestar a inflamación pode producir unha expresión aberrante dos xenes Nrf2 posteriores que induce a oncoxénese e resistencia á quimioterapia e/ou á radioterapia. Polo tanto, pódense desenvolver activadores altamente específicos de Nrf2 para minimizar os seus efectos pleiotrópicos. Varios activadores de Nrf2 demostraron unha mellora significativa das funcións antiinflamatorias nas enfermidades relacionadas co estrés oxidativo. O mellor exemplo de activador Nrf2 aprobado pola FDA e amplamente utilizado para o tratamento de enfermidades inflamatorias como a esclerose múltiple (EM) é o fumarato de dimetilo. Tecfidera (nome rexistrado de fumarato de dimetilo por Biogen) utilízase eficazmente para tratar as formas recurrentes de esclerose múltiple en gran número de pacientes [152]. Non obstante, a eficacia do uso de activadores de Nrf2 para tratar enfermidades inflamatorias require unha validación adicional para evitar os efectos nocivos de Nrf2. Polo tanto, o desenvolvemento de terapias para a actividade antiinflamatoria mediada por Nrf2 podería ter un impacto clínico significativo. Os estudos en curso sobre a vía de sinalización Nrf2 en todo o mundo dedícanse a desenvolver axentes terapéuticos altamente dirixidos para controlar os síntomas da inflamación e para previr e tratar o cancro, así como as enfermidades neurodexenerativas e outras enfermidades importantes.
En conclusión, Nrf2 detecta os niveis de estrés oxidativo no corpo humano e, en definitiva, axuda a promover a regulación das encimas e xenes antioxidantes e desintoxicantes. Debido a que a inflamación crónica causada por un aumento nos niveis de estrés oxidativo asociouse con enfermidades neurodegenerativas, Nrf2 pode desempeñar un papel esencial no tratamento de problemas de saúde como a enfermidade de Alzheimer, entre outros. O alcance da nosa información limítase aos problemas de saúde da columna vertebral e quiroprácticos. Para discutir o tema, non dubide en preguntar ao Dr. Jiménez ou en contacto connosco en�915-850-0900 .
Discusión temática adicional: aliviar a dor no xeonllo sen cirurxía
A dor no xeonllo é un síntoma ben coñecido que pode ocorrer debido a unha variedade de lesións e / ou afeccións no xeonllo, incluíndo lesións deportivas. O xeonllo é unha das articulacións máis complexas do corpo humano xa que está composto pola intersección de catro ósos, catro ligamentos, varios tendóns, dous meniscos e cartilaxe. Segundo a Academia Americana de Médicos de Familia, as causas máis comúns de dor no xeonllo inclúen a subluxación rotular, a tendinite rotuliana ou o xeonllo do saltador e a enfermidade de Osgood-Schlatter. Aínda que a dor no xeonllo é máis probable que se produza en persoas maiores de 60 anos, a dor no xeonllo tamén pode ocorrer en nenos e adolescentes. A dor no xeonllo pódese tratar na casa seguindo os métodos RICE, con todo, as lesións graves no xeonllo poden requirir atención médica inmediata, incluída a atención quiropráctica.
As enfermidades neurodexenerativas, como a enfermidade de Alzheimer e a enfermidade de Parkinson, afectan a millóns de individuos do mundo. Hai varias opcións de tratamento dispoñibles para tratar os síntomas de varias enfermidades neurodexenerativas, aínda que os resultados son a miúdo limitados. Os estudos de investigación atoparon que o estrés oxidativo causado por factores internos e externos pode ser unha causa para o desenvolvemento de enfermidades neurodexenerativas. O factor de transcrición, Nrf2, determinouse que funciona como un importante mecanismo de defensa contra o estrés oxidativo. O obxectivo do artigo seguinte é mostrar os efectos de Nrf2 sobre enfermidades neurodexenerativas.
Modulación da proteostase por factor de transcrición NRF2
As enfermidades neurodexenerativas están ligadas á acumulación de agregados proteicos específicos, o que suxire unha conexión íntima entre o cerebro ferido e a perda de proteostase. A proteostase refírese a todos os procesos polos que as células controlan a abundancia e dobra do proteoma grazas a unha ampla rede que integra a regulación das vías de sinalización, a expresión xénica e os sistemas de degradación de proteínas. Esta revisión intenta resumir os resultados máis relevantes sobre a modulación transcripcional da proteostase exercida polo factor de transcrición NRF2 (factor nuclear (eritróide derivado 2), como 2). NRF2 foi considerado clásicamente como o mestre regulador da resposta das células antioxidantes, aínda que actualmente está emerxendo como un compoñente clave da maquinaria de transdución para manter a proteostase. Como veremos, NRF2 podería ser concebido como un hub que recompila os sinais de emerxencia derivados da acumulación de proteínas mal plegadas para construír unha resposta transcricional coordinada e perdurable. Isto lógrase a través das funcións de NRF2 relacionadas co control de xenes implicados no mantemento da fisioloxía do retículo endoplasmático, o proteasoma e a autofagia.
O Factor Nuclear (2 eritróide derivado) como 2 (NRF2) é unha proteína básica-leucina-zíper considerada hoxe en día como un regulador mestre da homeostase celular. Controla a expresión basal e inducible por estrés de xenes 250 que comparten un potenciador de acción cis denominado elemento de resposta antioxidante (ARE) [1], [2], [3], [4], [5]. Estes xenes participan nas reaccións de desintoxicación de fase I, II e III, metabolismo do glutatión e peroxirredoxina / tiorredoxina, produción de NADPH a través da vía pentosa fosfato e enzima málica, oxidación de ácidos graxos, metabolismo do ferro e proteostase [6]. Dadas estas amplas funcións citoprotectoras, é posible que un único éxito farmacolóxico en NRF2 poida mitigar o efecto dos principais culpables de enfermidades crónicas, incluíndo o estrés oxidativo, inflamatorio e proteotóxico. O papel de NRF2 na modulación da defensa e resolución antioxidante da inflamación foi abordado en numerosos estudos (revisados en [7]). Aquí centraremos o seu papel na proteostase, é dicir, no control homeostático da síntese de proteínas, o pregamento, o tráfico e a degradación. Proporcionaranse exemplos no contexto de enfermidades neurodexenerativas.
A perda de proteostase inflúe na actividade de NRF2 en enfermidades neurodegenerativas
Unha característica xeral das enfermidades neurodexenerativas é a aparición de agregacións aberrantes dalgunhas proteínas. Así, os agregados de proteínas mal plegados de? -Sinucleína (? -SYN) atópanse na enfermidade de Parkinson (PD), placas? -Amiloides (A?) E enredos neurofibrilares TAU hiper-fosforilados na enfermidade de Alzheimer (AD), huntin (Htt) en A enfermidade de Huntington (HD), a superóxido dismutase 1 (SOD1) e a proteína de unión ao ADN TAR 43 (TDP-43) na esclerose lateral amiotrófica (ELA), a proteína priónica (PrP) en encefalopatías esponxiformes, etc. Os agregados de proteínas poden ter un impacto vías celulares, que á súa vez poden afectar os niveis e actividade de NRF2.
Diferentes capas de regulación Controlado na actividade de NRF2
En condicións fisiolóxicas, as células presentan baixos niveis de proteína NRF2 debido ao seu rápido volume de negocio. En resposta a diferentes estímulos, a proteína NRF2 acumúlase, entra no núcleo e aumenta a transcrición de xenes que conteñen ARE. Polo tanto, a xestión dos niveis de proteína NRF2 é un punto clave que debería integrar sinais de entrada positivos e negativos. Como discutiremos máis adiante, NRF2 é activado por diversos mecanismos de superposición para orquestrar unha resposta rápida e eficiente, pero por outra banda NRF2 podería inhibirse, probablemente nunha segunda fase, para desactivar a súa resposta.
Desde o punto de vista clásico, a activación de NRF2 considerouse como consecuencia da resposta celular a compostos oxidantes ou electrófilos. Neste sentido, o adaptador de ubiquitina E3 ligase proteína 1 asociada a ECH similar a Kelch (KEAP1) xoga un papel crucial. Os detalles moleculares serán tratados na sección 4.1. En resumo, KEAP1 actúa como un sensor redox debido a residuos críticos de cisteína que conducen á ubiquitinación NRF2 e degradación proteasómica. Ademais desta clásica modulación, NRF2 está profundamente regulado por eventos de sinalización. De feito, demostrouse que distintas quinasas fosforilan e regulan o NRF2. Por exemplo, o NRF2 pode fosforilarse mediante proteínas quinases activadas polo mitóxeno (MAPK), aínda que a súa contribución á actividade do NRF2 segue sen estar clara [8], [9], [10], [11]. A PKA quinasa e algúns isozimas PKC [12], CK2 [13] ou Fyn [14] fosforilan NRF2 modificando a súa estabilidade. O traballo anterior do noso grupo informou de que o glicóxeno sintase kinse-3? (GSK-3?) Inhibe o NRF2 por exclusión nuclear e degradación proteasómica [15], [25], [26], [27], [28], [29], [30]. Os detalles moleculares serán discutidos na sección 4.1. Ademais, NRF2 sométese a outros tipos de regulación. Por exemplo, a acetilación de NRF2 por CBP / p300 aumenta a súa actividade [17], mentres que está inhibida por miR153, miR27a, miR142-5p e miR144 [16], ou pola metilación das illas citosina-guanina (CG) dentro do promotor NRF2 [18].
Impacto dos agregados de proteínas nos mecanismos reguladores de NRF2
Nesta sección centraremos en como a acumulación de proteínas mal plegadas podería afectar á actividade de NRF2 proporcionando algúns dos camiños mencionados anteriormente como exemplos ilustrativos. En primeiro lugar, debemos considerar que a acumulación de proteínas estivo estreitamente ligada ao dano oxidativo. De feito, a acumulación e agregación de proteínas mal plegadas inducen a produción anormal de especies reactivas de osíxeno (ROS) a partir de mitocondrias e outras fontes [19]. Como se mencionou anteriormente, ROS modificará as cisteínas sensibles á redox de KEAP1 que levan á liberación, estabilización e localización nuclear de NRF2.
En canto ás proteínopatías, un exemplo de eventos de sinalización desregulados que poden afectar a NRF2 proporciónao a hiperactivación de GSK-3? en AD. GSK-3?, Tamén coñecida como TAU quinasa, participa na fosforilación desta proteína asociada a microtúbulos, resultando na súa agregación, formación de enredos neurofibrilares e interrupción do transporte axonal (revisado en [20]). Por outra banda, GSK-3? reduce drasticamente os niveis e a actividade de NRF2 como se mencionou anteriormente. Aínda que non está moi aceptada, a fervenza amiloide propón que o A tóxico? os oligómeros aumentan o GSK-3? actividade xunto coa hiper-fosforilación TAU e morte de neuronas [21], [22]. Hai diferentes modelos para explicar como A? favorece GSK3-? actividade. Por exemplo, A? únese ao receptor da insulina e inhibe as vías de sinalización PI3K e AKT, que son cruciais para manter o GSK-3? inactivado por fosforilación no seu residuo Ser9 N-terminal [23]. Por outra banda, extracelular A? interactúa cos receptores encrespados, bloqueando a sinalización WNT [24] e producindo de novo a liberación do GSK-3 activo ?. En resumo, A? a acumulación leva a unha hiperactivación anormal de GSK-3?, perturbando así unha resposta NRF2 adecuada.
Como se comenta na seguinte sección, as proteínas mal copiadas levan á activación de PERK e MAPKs, que á súa vez regulan a NRF2 [31], [8], [9], [10], [11]. Ademais, reportouse unha actividade disregulada de CBP / p300 en varias proteopatías [32] e tamén se mostrou unha diminución global da metilación do ADN nos cerebros AD [33], proporcionando razóns para explorar a relevancia destas conclusións na regulación NRF2.
Nós e outros observamos en necropsias de pacientes con AD e PD un aumento nos niveis de proteína NRF2 e algúns dos seus obxectivos, como a hemoxigenase 1 (HMOX1), NADPH quinona oxidase 1 (NQO1), p62, etc. por inmunohistoquímica [34], [35], [36], [37], [38], [39]. A regulación ascendente de NRF2 nestas enfermidades interprétase como un intento sen éxito do cerebro enfermo para recuperar valores homeostáticos. Non obstante, outro estudo indicou que NRF2 está predominantemente localizado no citoplasma das neuronas do hipocampo AD, o que suxire que a actividade transcricional de NRF2 é reducida no cerebro [40]. É concebible que a disparidade destas observacións estea relacionada cos cambios nos factores que controlan NRF2 ao longo das fases progresivas da neurodegeneración.
Tres sistemas principais contribúen á proteostase, é dicir, a resposta de proteínas despregadas (UPR), o sistema de proteasoma de ubiquitina (UPS) e a autofagia. A continuación, presentamos evidencias para visualizar NRF2 como un hub que conecta sinais de emerxencia activadas por agregados de proteínas coa maquinaria derivada de proteínas.
NRF2 participa na resposta de proteína despregada (UPR)
Activación de NRF2 en resposta ao UPR
O pregamento de proteínas oxidativas no ER está dirixido por varias vías distintas, a máis conservada das cales implica a proteína disulfuro-isomerase (PDI) e a sulfidriloxidasa endoplasmática oxidoreductina 1 (ERO1? E ERO1? En mamíferos) como doante de disulfuro. En breve, o PDI cataliza a formación e rotura de enlaces disulfuro entre os residuos de cisteína dentro das proteínas, ao dobrarse, debido á redución e oxidación dos seus propios aminoácidos de cisteína. O PDI recíclase coa acción do encima doméstico ERO1, que reintroduce os enlaces disulfuro no PDI [41]. O osíxeno molecular é o aceptor terminal de electróns de ERO1, que xera cantidades estequiométricas de peróxido de hidróxeno por cada enlace disulfuro producido [42]. As peroxidasas (PRX4) e as glutationas peroxidasas (GPX7 e GPX8) son encimas clave para reducir o peróxido de hidróxeno no ER. Cando este sistema oxido-redutor non funciona correctamente, prodúcese unha acumulación anormal de proteínas mal plegadas no ER e un conxunto de sinais chamadas resposta proteica despregada (EPU) transmítese ao citoplasma e ao núcleo para restablecer a homeostase do ER [43]. Identificáronse tres proteínas asociadas á membrana para detectar o estrés por ER nos eucariotas: factor de transcrición activante 6 (ATF6), ER eIF2 pancreático? quinasa (PERK, tamén proteína quinasa activada por ARN de dobre cadea, como a quinasa ER), e a quinasa que require inositol (IRE1). O dominio luminal de cada sensor está unido a unha chaperona de 1 kDa denominada proteína regulada pola glicosa (GRP78 / BIP). BIP disóciase ao estrés de ER para unir proteínas despregadas, o que leva á activación dos tres sensores [78].
NRF2 eo seu homólogo NRF1, tamén relacionados coa resposta antioxidante, participan na transdución do UPR no núcleo. No caso de NRF1, esta proteína está situada na membrana ER e sofre translocación nuclear tras a desglucosilación ou a escisión. Entón, a activación UPR leva ao procesamento de NRF1 e á acumulación nuclear do fragmento resultante no compartimento nuclear. Non obstante, a capacidade de transactivar os xenes que conteñen ARE deste fragmento NRF1 aínda está en discusión [45].
Glover-Cutter e compañeiros de traballo mostraron a activación do ortólogo NRF2 de C. elegans, SKN-1, con diferentes factores estressantes. A expresión aumentada de SKN-1 dependía de diferentes mediadores UPR, incluídos os ortólogos de gusanos IRE1 ou PERK [46]. Nas células con deficiencia de PERK, a síntese de proteínas deteriorada leva á acumulación de peróxidos endóxenos e á posterior apoptose [47]. O efector utilizado por PERK para protexer a ER a partir destes peróxidos podería ser NRF2, xa que se informou de que PERK fosforila NRF2 en Ser40, evitando así a súa degradación por KEAP1 [31]. A indución de ASK1 é tamén susceptible de desempeñar un papel nesta ruta a través da acción quinasa mediada por TRAF2 de IRE1 [48]. Aínda que o papel das MAPK na regulación de NRF2 aínda é controvertido, recentemente suxeriuse que a vía IRE1-TRAF2-ASK1-JNK podería activar NRF2 [49] (Fig. 1). Curiosamente, en C. elegans e células humanas, novas evidencias suxiren que a sulfenilación da cisteína de IRE1 quinase no seu circuíto de activación inhibe a UPR mediada por IRE1 e inicia unha resposta p38 antioxidante impulsada por NRF2. Os datos suxiren que IRE1 ten unha función antiga como centinela citoplasmática que activa p38 e NRF2 [50].
Figura 1 Regulación de NRF2 pola UPR. A acumulación de proteínas despregadas ou plegadas dentro do retículo endoplasmático pode iniciar a resposta de proteína despregada (UPR). En primeiro lugar, o chaperona BIP é liberado do dominio intraluminal dos sensores ER IRE1 e PERK para unirse ás proteínas desenvolvidas / mal plegadas. Isto permite a dimerización e trans-auto-fosforilación dos seus dominios citosólicos. A activación de PERK resulta en fosforilación directa de NRF2 en Ser40, o que leva á translocación de NRF2 ao núcleo e á activación dos xenes de destino. A activación de IRE1 induce o recrutamento de TRAF2 seguido da fosforilación e activación de ASK1 e JNK. Xa que se informou que JNK fosforila e activa NRF2, é razoable pensar que a activación de IRE1 levaría a unha maior actividade de NRF2.
Moitos estudos sobre a indución do EPU realizáronse co inhibidor da proteína tunicamicina glicosilación. O NRF2 parece ser esencial para a prevención da morte de células apoptóticas inducida pola tunicamicina [31] e a súa activación nestas condicións está motivada pola degradación autofáxica de KEAP1 [51]. En consecuencia, o silenciamento mediado por shRNA da expresión de NRF2 nas células β-TC-6, unha liña de célula β de insulinoma murino, aumentou significativamente a citotoxicidade inducida pola tunicamicina e provocou un aumento na expresión do marcador de estrés pro-apoptótico CHOP10. Por outra banda, a activación de NRF2 por 1,2-ditiol-3-tiona (D3T) reduciu a citotoxicidade da tunicamicina e atenuou a expresión de CHOP10 e PERK [52]. Curiosamente, as neuronas olfactivas sometidas a aplicación sistémica de tunicamicina aumentaron o NRF2 en paralelo con outros membros de UPR como CHOP, BIP, XBP1 [53]. Estes resultados ampliáronse a estudos in vivo, xa que a infusión ventricular lateral de tunicamicina en ratas induciu a expresión de PERK e NRF2 no hipocampo acompañados de déficits cognitivos significativos, aumento da fosforilación TAU e depósitos A? 42 [54].
NRF2 Up-Regulates Genes clave para o mantemento da fisioloxía ER
O lumen ER precisa unha abundante achega de GSH do citosol para manter a química do disulfuro. NRF2 modula encimas cruciais do metabolismo de GSH no cerebro, como o transporte de cistina / glutamato, β-glutamato cisteína sintetase (? -GS), subunidades catalíticas de glutamato-cisteína ligasa e modulador (GCLC e GCLM), glutatión redutasa (GR) e glutatión peroxidasa (GPX) (revisado en [55]). A relevancia de NRF2 no mantemento de GSH no ER está avalada polo descubrimento de que a activación farmacolóxica ou xenética de NRF2 resulta nunha maior síntese de GSH a través de GCLC / GCLM, mentres que a inhibición da expresión destes encimas por derrubamento de NRF2 causou unha acumulación de danos. proteínas dentro do ER levando á activación do UPR [56].
En C. elegans hai varios compoñentes dos xenes obxectivo UPR regulados por SKN-1, incluíndo Ire1, Xbp1 e Atf6. Aínda que NRF2 regula a expresión de varios xenes peroxidasa (PRX) e glutatión peroxidasa (GPX) en mamíferos (revisado en [57]), só GPX8 é un encima localizado ER de boa fe, que alberga o sinal de recuperación de KDEL [58]. A perda de GPX8 provoca a activación de UPR, a fuga de peróxido de hidróxeno derivado de ERO1? Ao citosol e a morte celular. Peróxido de hidróxeno derivado de ERO1? a actividade non pode difundirse do ER ao citosol debido á acción concertada de GPX8 e PRX4 [59]. Neste sentido, unha análise da vía de defensa antioxidante-matriz de expresión xénica usando ARN de tecido de rato de tipo salvaxe e NRF2-nulo, revelou que a expresión de GPX8 estaba regulada á baixa en ausencia de NRF2 [60]. En liña con isto, a análise do transcriptoma de mostras de pacientes que padecen neoplasias mieloproliferativas, policitemia ou mielofibrosis, as enfermidades asociadas tamén ao estrés oxidativo e á inflamación crónica de baixo grao, mostran niveis de expresión máis baixos tanto de NRF2 como de GPX8 en comparación cos suxeitos control [61]. Aínda non hai estudos que impliquen especificamente GPX8 na protección do cerebro humano, pero unha análise do transcriptoma en ratos indica un aumento compensatorio do GPX8 en resposta á toxina parkinsónica MPTP [62].
Impacto de NRF2 na desregulación UPR en enfermidades neurodegenerativas
Posteriormente, o mal funcionamento dos encimas PDI e a activación crónica da UPR poderían iniciar ou acelerar a neurodegeneración. As neuronas afectadas polas enfermidades, os modelos animais de enfermidades neurodexenerativas e os tecidos humanos post mortem evidencian a regulación de varios marcadores UPR na maioría destes trastornos. A alteración da vía PDI / UPR nas enfermidades neurodexenerativas foi ben revisada en [63] pero deben considerarse os seguintes aspectos das mostras post-mortem do cerebro. Os niveis de PDI aumentan en neuronas con enredos e en corpos Lewy de pacientes con AD e PD, respectivamente [64], [65]. Os PDI e ERP57 están regulados no CSF por pacientes con ALS e nos cerebros dos suxeitos CJD [66], [67], [68]. BIP, PERK, IRE1 e ATF6 son elevados en mostras de pacientes con AD, PD ou ALS [69], [70], [71], [67]. BIP, CHOP e XBP1 son elevados nas mostras cerebrais post-mortem do HD [72], [73]. Ademais, atopouse unha regulación ascendente de ERP57, GRP94 e BIP en tecidos de cortiza de pacientes de CJD [74]. En conxunto, esta evidencia revela que a acumulación de proteínas mal plegadas no parénquima cerebral leva a unha activación deletérea e crónica da EPU. Curiosamente, hai un estudo recente que une a activación de NRF2 polo PERK no inicio da AD. Neste estudo, os autores analizaron se os cambios mediados por estrés oxidativo en NRF2 e UPR poden constituír os primeiros eventos na patoxénese AD usando células sanguíneas periféricas humanas e un modelo de rato transxénico AD en diferentes etapas da enfermidade. O aumento do estrés oxidativo e o aumento do pSer40-NRF2 observáronse en células mononucleares de sangue periférico humano illadas de individuos con deterioro cognitivo leve. Ademais, informaron de homeostase de calcio en ER e marcadores de estrés ER regulados por riba nesas células a partir de individuos con deterioro cognitivo lixeiro e leve AD [75].
Regulación mutua de NRF2 e do sistema de proteasoma de ubiquitina (UPS)
O UPS modula os niveis de proteína NRF2
A UPS participa na degradación de proteínas danadas ou mal plegadas e controla os niveis de moléculas reguladoras clave no citosol e no núcleo. O núcleo central deste sistema é unha gran enzima multisubunita que contén un complexo activo proteolítico chamado 20S. O núcleo proteasoma de 20S degrada as proteínas desenvolvidas, pero a unión a distintos complexos proteicos reguladores cambia a súa especificidade e actividade. Por exemplo, a adición dunha ou dúas subunidades reguladoras 19S ao núcleo 20S constitúe o proteasoma 26S e cambia a súa especificidade cara ás proteínas dobradas nativas [76], [77]. A degradación do proteasomal precisa da unión covalente da ubiquitina. A conxugación de ubiquitina procede mediante un mecanismo en cascada de tres pasos. En primeiro lugar, o encima activador de ubiquitina E1 activa a ubiquitina nunha reacción que require ATP. Entón, un encima E2 (proteína portadora de ubiquitina ou enzima conxugante de ubiquitina) transfire a ubiquitina activada de E1 ao substrato que está específicamente ligado a un membro da familia de ligas de proteína ubiquitina, chamada E3. Aínda que o destino exacto da proteína ubiquitinada dependerá da natureza da cadea de ubiquitina, este proceso xeralmente resulta na degradación do proteasoma 26S [78].
O E3-ligase KEAP1 é o inhibidor máis coñecido de NRF2. O mecanismo da regulación KEAP1 explica de xeito elegante como os niveis de NRF2 axústanse ás fluctuacións dos oxidantes. En condicións basais, o NRF2 recén sintetizado é agarrado polo homodímero KEAP1, que une unha molécula NRF2 en dúas secuencias de aminoácidos con afinidade baixa (aspartato, leucina, glicina; DLG) e alta (glutamato, treonina, glicina, glutamato; ETGE). A interacción con KEAP1 axuda a presentar NRF2 ao complexo de proteínas CULLIN3 / RBX1, o que resulta na súa ubiquitinación e posterior degradación do proteasomal. Non obstante, a modificación redox de KEAP1 impide a presentación de NRF2 ao UPS representado por CULLIN3 / RBX1. Como resultado, NRF2 recén sintetizado escapa á degradación dependente de KEAP1, acumúlase no núcleo e activa xenes que conteñen ARE [79], [80], [81], [82].
O adaptador E3-ligasa? -TrCP tamén é un homodímero que participa nos eventos de sinalización relacionados coa fosforilación de NRF2 por GSK-3 ?. Esta quinase fosforila residuos específicos de serina de NRF2 (aspartato, serina, glicina, serina isoleucina; DSGIS) para crear un dominio de degradación que logo é recoñecido por? -TrCP e marcado para a degradación do proteasoma por un complexo CULLIN1 / RBX1. A identificación dos aminoácidos específicos fosforilados por GSK-3? neste degron levouse a cabo mediante unha combinación de mutaxénese dirixida ao sitio do dominio Neh6, electroforese en xel 2D [15], [26] e espectroscopia de masas [83]. En consecuencia, a inhibición de GSK-3? por medicamentos ou siRNA altamente selectivos contra as isoformas GSK-3 provocaron un aumento dos niveis de proteína NRF2. Resultados similares atopáronse con siRNAs contra as isoformas 1 e 2.? -TrCP Estabilización de NRF2 tras GSK-3? a inhibición produciuse en fibroblastos de embrión de rato con déficit de KEAP1 e nun mutante de supresión NRF2 ectópicamente carente dos residuos críticos de ETGE para a unión de alta afinidade a KEAP1, demostrando ademais unha regulación independente de KEAP1.
No contexto das enfermidades neurodexenerativas, podemos imaxinar a modulación de NRF2 polo SAI de dous xeitos diferentes. Por unha banda, o sistema KEAP1 sentiría un desequilibrio redox derivado da acumulación de proteínas mal plegada, mentres que o eixe GSK-3 /? - TrCP actuaría como un participante activo na transducción de sinalización alterada pola perda de proteóstase (Fig. 2).
Figura 2 O UPS controla os niveis de NRF2. En condicións homeostáticas, a acción dos adaptadores de ligasas E2 KEAP3 e? -TrCP manteñen niveis baixos de NRF1. Á esquerda, NRF2 únese aos dominios Kelch dun homodímero KEAP1 a través de motivos de afinidade baixa (DLG) e alta (ETGE). A través do seu dominio BTB, KEAP1 únese simultaneamente a un complexo CULLIN3 / RBX1, o que permite a ubiquitinación e degradación NRF2 polo proteasoma 26 S. Ademais, GSK-3? fosforila os residuos Ser335 e Ser338 de NRF2 para crear un dominio de degradación (DpSGIpSL) que logo é recoñecido polo adaptador de ubiquitina ligase? -TrCP e marcado para a degradación do proteasoma por un complexo CULLIN3 / RBX1. Á dereita, despois da exposición a especies reactivas de osíxeno ou electrófilos, modifícanse os residuos críticos de Cys en KEAP1, o que fai que KEAP1 non poida interactuar de xeito eficiente con NRF2 ou CULLIN3 / RBX1 e entón este factor de transcrición aumenta a súa vida media e a súa actividade transcricional cara aos xenes ARE. As vías de sinalización que provocan a inhibición de GSK-3?, Tal fosforilación AKT en Ser9, provocan unha degradación deteriorada por NRF2 polo proteasoma, a acumulación e a indución de xenes diana.
NRF2 aumenta a actividade de UPS a través do control de transcrición da subunidade do proteasoma
NRF2 regula a expresión de varias subunidades do proteasoma, protexendo así a célula da acumulación de proteínas tóxicas. Vinte xenes relacionados co proteasoma e a ubiquitinación parecen estar regulados por NRF2, segundo unha ampla análise de microarrays a partir de ARN hepático que se creou co indutor de NRF2 D3T [84]. Nun estudo posterior, os mesmos autores evidenciaron que a expresión da maioría das subunidades do proteasoma 26S melloráronse ata tres veces en fígados de ratos tratados con D3T. Os niveis de proteínas das subunidades e a actividade dos proteasomas aumentaron coordinadamente. Non obstante, non se observou ningunha indución en ratos nos que se alterou o factor de transcrición NRF2. A actividade promotora da subunidade do proteasoma PSMB5 (20S) aumentou coa sobreexpresión de NRF2 ou co tratamento con activadores en fibroblastos embrionarios de rato e identificáronse ARE no promotor proximal de PSMB5 [85]. A activación farmacolóxica de NRF2 deu lugar a niveis elevados de expresión de subunidades representativas do proteasoma (PSMA3, PSMA6, PSMB1 e PSMB5) só en fibroblastos humanos non senescentes que conteñen NRF2 funcional [86]. A activación de NRF2 durante a adaptación ao estrés oxidativo resulta nunha alta expresión do PSMB1 (20S) e do PA28? subunidades (ou S11, regulador do proteasoma) [87]. Ademais, os resultados das células nai embrionarias humanas revelaron que NRF2 controla a expresión da proteína de maduración do proteasoma (POMP), unha chaperona proteasoma, que á súa vez modula a proliferación de células nai embrionarias humanas auto-renovables, a diferenciación de tres capas xerminais e a reprogramación celular [ 88]. En conxunto, estes estudos indican que NRF2 regula a expresión dos compoñentes clave do SAI e, polo tanto, contribúe activamente á eliminación de proteínas que doutro xeito serían tóxicas.
O eixe NRF2-UPS en enfermidades neurodexenerativas
O papel do SAI nas enfermidades neurodexenerativas é un campo de intenso debate. Os estudos iniciais informaron diminuír a actividade do proteasoma nas necropsias humanas de pacientes afectados por varias enfermidades neurodexenerativas. Non obstante, outros estudos que empregan enfoques in vitro e in vivo atoparon unha actividade proteasoma inalterada ou incluso aumentada (revisada en [89]). Unha posible explicación desta discrepancia é que os niveis dos compoñentes do SAI poden cambiar durante a progresión da enfermidade e en diferentes rexións cerebrais como se suxeriu para os obxectivos de NRF2.
A pesar desta controversia, débese notar que a regulación ascendente dos xenes proteasomos que conteñen ARE reforzará o SAI aumentando a depuración de proteínas tóxicas no cerebro. De feito, a ablación de NRF1, tamén moduladora da resposta antioxidante, nas células neuronais leva a unha diminución da actividade do proteasoma e da neurodexeneración. Experimentos de inmunoprecipitación de cromatina e análise transcricional demostraron que PSMB6 está regulado por NRF1. Ademais, o perfil de expresión xénica levou á identificación de NRF1 como un regulador transcricional clave dos xenes do proteasoma nas neuronas, o que suxire que as perturbacións en NRF1 poden contribuír á patoxénese das enfermidades neurodexenerativas [90]. Curiosamente, NRF1 eo seu longo isoformo chamado TCF11 mostraron que regulan xenes proteasomas que conteñen ARE sobre a inhibición do proteasoma nun circuíto de realimentación para compensar a actividade proteolítica reducida [91], [92].
En relación a NRF2, hai unha correlación entre a redución dos niveis de NRF2, RPT6 (19 S) e PSMB5 (20 S) no cerebro medio dos ratos deficientes de DJ-1 tratados coa neurotoxina paraquat [93]. Ademais, o composto natural sulforaphane (SFN) dá unha imaxe máis robusta de NRF2 como un modulador crucial do SAI. Experimentos in vitro con células de neuroblastoma murino Neuro2A evidencian unha maior expresión das subunidades catalíticas do proteasoma e das súas actividades de peptidasa en resposta a SFN. Esta droga protexe as células da citotoxicidade mediada polo peróxido de hidróxeno e da oxidación das proteínas de forma dependente da función do proteasoma [94]. Ademais, Liu e compañeiros de traballo contrataron a un ratón reporteiro para supervisar a actividade do SAI en resposta a SFN no cerebro. Estes ratos expresan de xeito omnipresente a proteína de fluorescencia verde (GFP) fusionada a un sinal de degradación constitutiva que promove a súa rápida degradación polo UPS (GFPu). No córtex cerebral, SFN reduciu o nivel de GFPu cun incremento paralelo nas actividades de tipo chemotripsina (PSMB5), caspase (PSMB2) e tipo tripsina (PSMB1) do proteasoma 20 S. Ademais, o tratamento das células derivadas de Huntington con SFN revelou que a activación de NRF2 mellorou a degradación do mHtt e reduciu a citotoxicidade de mHtt [95]. O mecanismo principal da acción de SFN é a través da inducción de NRF2 [96]. A contribución específica de NRF2 debería tratarse empregando sistemas nulos de NRF2 en estudos posteriores.
Conexión funcional entre NRF2 e Macroautophagy
Os niveis de proteína NRF2 modúlanse pola proteína adaptadora P62
A autofaxia refírese á degradación de compoñentes citosólicos dentro dos lisosomas. Este proceso úsase para a depuración de proteínas de longa duración e plegadas, así como de orgánulos danados. Un enlace directo entre NRF2 e autofagia observouse por primeira vez en conexión coa proteína adaptadora p62, tamén denominada SQSTM1 [97], [98], [99], [100], [101]. Esta proteína transporta proteínas ubiquitinadas ás máquinas de degradación do proteasoma e lisosomas e secuestra proteínas danadas en agregados antes da súa degradación. P62 presenta un dominio asociado á ubiquitina (UBA), para a unión a proteínas ubiquitinadas, e unha rexión que interactúa con LC3 (LIR) para a integración coa membrana autofagosoma a través do receptor de autofagia LC3.
Aínda que a indución mediada por p62 de NRF2 e os seus xenes diana informouse por primeira vez en 2007 [102], o mecanismo molecular non se comprendeu completamente ata o descubrimento da súa interacción con KEAP1 [103], [98], [99], [100] ], [101]. Komatsu e compañeiros de traballo identificaron unha rexión de interacción KEAP1 (KIR) en p62 que unía a KEAP1 no mesmo peto de superficie básico que NRF2 e cunha afinidade de unión similar ao motivo ETGE en NRF2, o que suxire a competencia entre p62 e NRF2. A fosforilación de Ser351 no motivo KIR en p62 (349-DPSTGE-354) demostrou aumentar a súa afinidade por KEAP1, competindo coa unión a NRF2 e permitindo a súa acumulación e activación transcricional dos seus xenes diana [98], [99]. De feito, a sobreexpresión de p62 provocou unha reducida ubiquitinación de NRF2 e a conseguinte estabilización, así como a indución dos seus xenes obxectivo [104]. Suxeriuse que algunhas quinasas participan na fosforilación de p62. O obxectivo dos mamíferos do complexo 1 de rapamicina (mTORC1) pode estar implicado, xa que o tratamento co inhibidor do mTOR rapamicina suprimiu a fosforilación de p62 e a regulación descendente de KEAP1 ao tratar o arsenito. Recentemente, demostrouse que a quinasa 1 activada por TGF -? - (TAK1) tamén podería fosforilar p62, mellorando a degradación de KEAP1 e a regulación de NRF2. Os autores deste estudo suxiren que este é un xeito de regular a redoxtasis celular en condicións de estado estacionario, xa que a deficiencia de TAK1 regula a ROS á falta de oxidante esóxeno en diferentes tecidos de rato en paralelo cunha redución dos niveis de proteína NRF2 [105 ].
Un constructor p62 sen o dominio UBA aínda era capaz de unir a KEAP1, o que implica que a interacción non dependía de ubiquitinated KEAP1 [101]. Non obstante, o homólogo p62 en Drosophila melanogaster, chamado Ref (2), non contén un motivo KIR e non interactúa directamente con DmKEAP1, aínda que pode unirse a DmKEAP1 ubiquitinado a través do dominio UBA. Ademais, DmKEAP1 pode interactuar directamente con Atg8 (homólogo a LC3 de mamíferos). A deficiencia de KEAP1 resulta en Atg8 e indución autofagia dependente do ortólogo NRF2 CncC e independente en TFEB / MITF [106]. A relación entre NRF2 e autofagia parece conservarse, destacando a súa relevancia funcional.
A indución de NRF2 por p62 é o resultado tanto da competencia para unirse a KEAP1 como á degradación de KEAP1 no lisosoma. O silenciamento de p62 con siRNA duplicou a semivida de KEAP1 en paralelo cunha diminución de NRF2 e os seus xenes obxectivo [101]. De acordo, a ablación da expresión p62 evidenciou un aumento dos niveis de KEAP1 en comparación cos ratos de tipo salvaxe. Moi relevante, o incremento nos niveis de KEAP1 non foi afectado por inhibidores do proteasoma, pero reduciuse baixo a autofagia que provoca a fame [107]. De feito, KEAP1 está presente en células de mamíferos en vesículas autofáxicas decoradas con p62 e LC3 [99], [100], [103]. Todos estes datos suxiren que KEAP1 é un substrato da maquinaria de macroautofaxia, pero este problema debería analizarse con máis detalle debido á existencia dalgúns resultados controvertidos. Os niveis de proteína KEAP1 incrementáronse nos ratos nulos de Atg7, un efector clave da macroautofaxia [107], pero a inhibición farmacolóxica da macroautofaxia con torin1, E64 / pepstatin ou bafilomicina non puido acumular KEAP1 [107], [100]. En xeral, estes resultados suxiren que o aumento dos niveis de p62 secuencia KEAP1 en vacúolos autofáxicos e probablemente estes resultados na degradación autofágica de KEAP1 permitindo a activación de NRF2 (Fig. 3). Dous estudos diferentes informaron de que o ácido sulfínico redutasa SESTRINS desempeña un papel importante neste contexto. SESTRIN 2 interactúa con p62, KEAP1 e RBX1 e facilita a degradación dependente de p62 da activación de xenes de destino KEAP1 e NRF2 [108]. Outro estudo mostrou que SESTRIN 2 interactúa con ULK1 e p62, promovendo a fosforilación de p62 en Ser403, o que facilitou a degradación das proteínas de carga, incluíndo KEAP1 [109].
Figura Os niveis de 3 NRF2 están regulados pola proteína adaptadora p62. A fosforilación de Ser 351 no motivo KIR de p62 (349-DPSTGE-354) por mTORC1, TAK1 ou outras quinasas resulta nunha maior afinidade por unirse a KEAP1 debido á semellanza co motivo ETGE en NRF2. Como consecuencia, a p62 fosforilada despraza a NRF2 e únese a KEAP1. O motivo LIR en p62 permite a interacción con LC3 na membrana autofagosómica, polo que o complexo p62-KEAP1 é finalmente degradado no lisosoma. Como consecuencia, NRF2 é capaz de acumularse, trasladarse ao núcleo e aumentar a transcrición de xenes que conteñen ARE, incluíndo p62. Este mecanismo regulador proporciona unha resposta NRF2 perdurable, xa que KEAP1 ten que ser recientemente sintetizado para inhibir a actividade de NRF2.
Modulación de xenes de macroautofagia por NRF2
NRF2 regula a expresión dos xenes relevantes para a macroautofaxia, así como para a UPR ea UPS. As primeiras evidencias procedían de estudos nos que se mostrou que a expresión p62 foi inducida por exposición a electrófilos, ROS e óxido nítrico [110], [111], [112]. O mecanismo de inducción foi descrito algúns anos despois co descubrimento de que p62 contén un ARE funcional no seu xene promotor [99]. Nun estudo recente, atopáronse e validáronse varias AREs funcionais tras análises bioinformáticos e ensaios de ChIP. Ademais, os fibroblastos embrionarios do rato e as neuronas corticales de ratos eliminatorios de Nrf2 mostraron unha expresión reducida de p62, que podería ser rescatada cun lentivirus que expresa NRF2. Do mesmo xeito, a deficiencia de NRF2 reduciu os niveis de p62 en neuronas feridas de ratones hipocampo [36]. Polo tanto, suxeriuse que a activación de NRF2 aumenta os niveis de p62, resultando na degradación de KEAP1 e favorecendo aínda máis a estabilización de NRF2 nun circuíto de feedback positivo. Este mecanismo non canónico da inducción de NRF2 require cambios na expresión xénica e pode ser unha resposta relevante ao estrés celular prolongado.
A proteína de recoñecemento de carga NDP52 foi regulada transcricionalmente por NRF2. NDP52 funciona de forma similar a p62, recoñecendo as proteínas ubiquitinadas e interactuando con LC3 a través dun dominio LIR, de xeito que as cargas son degradadas nos lisosomas. Atopáronse cinco AREs supostas na secuencia de ADN do promotor Ndp52. Tres deles identificáronse con diferentes construcións mutantes e ensaios ChIP como indispensables para a transcrición de Ndp2 mediada por NRF52 [113]. Nótese que os niveis de ARNm de Ndp52 reducíronse no hipocampo dos ratos Nrf2-knockout. Unha destas secuencias tamén foi validada nun estudo independente como un ARE [2] regulado por NRF36.
Non obstante, o papel de NRF2 na modulación da autofagia non se limita á inducción destas dúas proteínas de recoñecemento de carga. Co fin de obter unha visión máis profunda do papel de NRF2 na modulación de xenes relacionados coa autofaxia adicional, o noso grupo analizou a base de datos de inmunoprecipitación de cromatina ENCODE para dúas proteínas, MAFK e BACH1, que se unen ás AREs reguladas por NRF2. Empregando un script xerado a partir da consecuencia do consenso ARE de JASPAR, identificamos varias AREs supostas en moitos xenes autofagistas. Doce destas secuencias validáronse como os AREs regulados por NRF2 en nove xenes de autofagia, cuxa expresión diminuíu nos fibroblastos de embrións de rato de ratos Nrf2-knockout pero podían ser restaurados por un lentivirus que expresaba NRF2. O noso estudo demostrou que NRF2 activa a expresión dalgúns xenes implicados en diferentes etapas do proceso autofágico, incluíndo a iniciación da autofagia (ULK1), o recoñecemento de carga (p62 e NDP52), formación autofagosoma (ATG4D, ATG7 e GABARAPL1), alongamento (ATG2B e ATG5) ), e autorización de autoliseosoma (ATG4D). En consecuencia, o fluxo de autofagia en resposta ao peróxido de hidróxeno foi afectado cando NRF2 estaba ausente [36].
Relevancia da expresión dos xenes de macroautofábrica mediada por NRF2 en trastornos neurodexenerativos
A autofagia defectuosa demostrou que desempeña un papel importante en varias enfermidades neurodexenerativas [114] e a ablación de autofagia leva á neurodegeneración en ratos [115], [116]. Os ratos knockout de Atg7 revelaron que a deficiencia de autofagia resulta nunha acumulación de p62 en corpos de inclusión positivos en ubiquitina. KEAP1 foi secuestrado nestes corpos de inclusión, dando lugar a estabilización de NRF2 e indución de xenes obxectivo [103]. É importante destacar que se identificou unha acumulación excesiva de p62 xunto con proteínas ubiquitinadas en enfermidades neurodegenerativas, incluíndo AD, PD e ALS [117]. De feito, as neuronas que expresan altos niveis de APP ou TAU de pacientes con AD tamén expresaron p62 e NRF2 nuclear, o que suxire o seu intento de degradar agregados intraneuronais a través da autofagia [36].
A deficiencia de NRF2 agrava a agregación de proteínas no contexto de AD. De feito, aumentaron os niveis de TAU fosforilado e insoluble en sarkosil en ratos Nrf2-knockout, aínda que non se puido detectar ningunha diferenza nas actividades de quinasa ou fosfatase en comparación co fondo de tipo salvaxe [113]. É importante destacar que NDP52 demostrou co-localizarse con TAU en neuronas murinas e a interacción directa entre fosfo-TAU e NDP52 demostrouse mediante experimentos de co-inmunoprecipitación tanto en ratos como en mostras de AD, apuntando ao seu papel na degradación de TAU. Curiosamente, o silencio de NDP52, p62 ou NRF2 nas neuronas provocou un aumento do fosfo-TAU [113], [118]. Ademais, atopáronse maiores agregados intraneuronais de APP no hipocampo dos ratos APP / PS1? E9 cando estaba ausente NRF2. Isto correlacionouse con marcadores de autofaxia alterados, incluíndo un aumento das relacións fosfo-mTOR / mTOR e fosfo-p70S6k / p70S6k (indicativo da inhibición da autofaxia), niveis aumentados de pre-catepsina D e un maior número de corpos multivesiculares [119]. En ratos que coexpresan APP humana (V717I) e TAU (P301L), a deficiencia de NRF2 provocou un aumento dos niveis de total e de fosfo-TAU na fracción insoluble e un aumento de agregados intraneuronais de APP, xunto con niveis neuronais reducidos de p62, NDP52, ULK1, ATG5 e GABARAPL1. A co-localización entre a proteína adaptadora p62 e APP ou TAU reduciuse en ausencia de NRF2 [36]. En xeral, estes resultados destacan a importancia de NRF2 na autofaxia neuronal.
Diferentes factores de transcrición actúan de xeito coordinado para modificar a proteostase
En condicións de estado estacionario, a proteostase contrólase a través de interaccións proteína-proteína e modificacións postraducionais obtendo unha resposta rápida. Non obstante, a adaptación celular require a regulación transcripcional dos xenes UPR, UPS e autofagia. Considerando que as células nerviosas están sometidas continuamente a insultos tóxicos de baixo grao, incluíndo o estrés oxidativo e proteotóxico, un reforzo da proteostase inducida pola modulación transcricional pode axudar a previr a dexeneración cerebral.
No caso do UPR, a activación de cada un dos tres brazos resultará finalmente na inducción transcricional de certos xenes (revisados en [43]). Por exemplo, un fragmento derivado de ATF6 (ATF6f) únese a elementos de resposta de estrés ER (ERSE) e induce a expresión de varios xenes, incluíndo XBPI, BIP e CHOP. Ademais, a sinalización de PERK leva á activación do factor de transcrición ATF4, que controla a expresión de múltiples xenes relacionados coa UPR e algúns outros incluíndo os xenes de destino NRF2 Hmox1 e p62. Finalmente, a activación de IRE1 resulta na xeración dun factor de transcrición activo, XBP1 empalmado (XBP1s), que controla a transcrición de xenes que codifican proteínas implicadas no pregamento das proteínas.
Por outra banda, demostrouse que NRF1 é necesaria para a expresión do xene proteasomal no cerebro, xa que os ratos Nrf1-knockout exhibiron unha expresión reducida de xenes que codifican varias subunidades do núcleo 20S, así como o complexo regulador 19S xunto coa función proteasomal alterada [90 ]. Tanto NRF1 como NRF2 únense a secuencias de ARE nas rexións promotoras dos seus xenes obxectivo, o que suxire que teñen actividades de transcrición superpuestas, aínda que difiren nos seus mecanismos reguladores e na localización celular [120].
Os factores de transcrición da familia Forkhead box O (FOXO) controlan a expresión de múltiples xenes relacionados coa autofagia. Similar ao que ocorre con NRF2, hai varias capas de regulación da actividade dos membros da FOXO, que poden ser inducidas por estrés nutricional ou oxidativo [121]. Finalmente, o factor de transcrición TFEB, considerado o principal regulador da biogênese lisosomal, xoga un papel crucial na regulación da autofagia en condicións de estrés nutricional. Así, a inhibición de mTORC1 leva á translocación nuclear de TFEB e á inducción da expresión de xenes autofagistas [122].
En xeral, a existencia de diferentes reguladores de transcrición destes mecanismos tamén suxire a diafonía e mecanismos parcialmente redundantes que poden asegurar a proteostase en diferentes circunstancias. En consecuencia, NRF2 pode ter un papel relevante nos tecidos que soportan altos niveis de estrés oxidativo. Por exemplo, NRF2 inducido polo estrés oxidativo pode funcionar baixo condicións ricas en nutrientes para a autofaxia transcricionalmente reguladora, similar ao atopado para TFEB en condicións de inanición. Ademais, o cerebro funciona en gran parte baixo condicións ricas en nutrientes, o que pon a NRF2 como un mecanismo relevante para activar a autofagia nas neuronas.
Potencial terapéutico prometedor para NRF2 en proteinopatías
Nos últimos anos logrouse un gran avance no coñecemento das funcións reguladoras do EPU, UPS e autofagia na actividade de NRF2, así como a transcrición recíproca mediada por NRF2 de compoñentes destes tres sistemas. Polo tanto, poden xurdir novas posibilidades terapéuticas baseadas na explotación de NRF2 como regulador crucial da eliminación de proteínas nas enfermidades neurodegenerativas.
Non obstante, unha pregunta clave restante é se será útil ou perjudicial aumentar os niveis de NRF2 no cerebro. A análise de datos epidemiolóxicos pode proporcionar unha resposta parcial, xa que indica que o xene NFE2L2 é altamente polimórfico e algúns polimorfismos dun só nucleótido atopados na súa rexión reguladora do promotor poden proporcionar un rango de variabilidade fisiolóxica na expresión xénica a nivel de poboación e algúns haplotipos. asociáronse cunha diminución do risco e / ou un inicio retardado de AD, PD ou ELA [123]. Ademais, como discutiron Hayes e colegas [124], o efecto NRF2 podería ter unha resposta en forma de U, o que significa que niveis demasiado baixos de NRF2 poden provocar unha perda de citoprotección e unha maior susceptibilidade aos estresantes, mentres que demasiado NRF2 pode perturbar o equilibrio homeostático cara a un escenario redutivo (estrés redutivo), que favorecería o plegamento e a agregación de proteínas. Os baixos niveis de NRF2 no cerebro apoian a idea de que unha lixeira regulación ascendente pode ser suficiente para lograr un beneficio en condicións patolóxicas. De feito, o papel protector da activación farmacolóxica mediada por NRF2 da depuración de proteínas demostrouse en diferentes modelos de cultivo celular de neurodexeneración e in vivo.
SFN é un activador farmacolóxico de NRF2 que demostrou inducir a expresión xénica proteasómica e autofaxia [95], [36]. Curiosamente, Jo e colegas demostraron que SFN reduciu os niveis de TAU fosforilado e aumentou Beclin-1 e LC3-II, suxerindo que a activación de NRF2 pode facilitar a degradación desta proteína tóxica a través da autofaxia [113]. Ademais, a degradación de mHtt mellorouse con SFN, e reverteuse co uso de MG132, o que indica degradación proteasómica desta proteína tóxica [95]. Informouse dunha degradación mediada por autofaxia de TAU fosfo e insoluble coa fisetina flavonoide orgánica. Este composto foi capaz de inducir a autofaxia promovendo simultaneamente a activación e translocación nuclear de TFEB e NRF2, xunto con algúns dos seus xenes diana. Esta resposta evitouse por silenciar TFEB ou NRF2 [125]. Bott e colegas informaron de efectos beneficiosos dun activador simultáneo de NRF2, NRF1 e HSF1 sobre a toxicidade das proteínas na atrofia muscular espinal e bulbar, un trastorno neurodexenerativo causado pola expansión das repeticións de CAG que codifican poliglutamina na que están presentes os agregados de proteínas [126]. O potencial da activación de NRF2 para o tratamento de trastornos neurodexenerativos demostrouse coa aprobación de BG-12, a formulación oral do dimetil fumarato (DMF) indutor de NRF2, para o tratamento da esclerose múltiple [127], [128]. O éxito do DMF con enfermidades autoinmunes cun forte compoñente inflamatorio suxire que as enfermidades neurodexenerativas poden beneficiarse da reposición deste medicamento. Nun recente estudo preclínico dun modelo de β-sinucleinopatía de PD, demostrouse que o DMF era neuroprotector debido, en parte, á súa indución de autofaxia [129]. Os estudos que reportan efectos beneficiosos de NRF2 sobre a neurodexeneración pero que non se centran no seu efecto sobre a depuración de proteínas son aínda máis abundantes (para unha revisión completa, ver [7]). Isto é bastante relevante, xa que destaca os múltiples procesos daniños que poden ser dirixidos simultaneamente por un único golpe en NRF2, incluíndo tamén o estrés oxidativo, a neuroinflamación ou a disfunción mitocondrial. Non obstante, será necesario traballar no futuro para determinar definitivamente se a activación farmacolóxica de NRF2 pode ser unha estratexia válida para facilitar a degradación de proteínas tóxicas no cerebro.
Como se explicou antes, exacerbouse GSK-3? informouse de actividade en enfermidades neurodexenerativas e especulouse que a consecuente redución de NRF2 pode ser parcialmente responsable do resultado prexudicial. Nestas condicións patolóxicas, os inhibidores de GSK-3 tamén poderían cooperar para aumentar os niveis de NRF2 e a proteóstase. Os efectos beneficiosos dos inhibidores de GSK-3 foron informados en diferentes modelos de neurodexeneración e, máis interesante, demostrouse que a represión GSK-3 reduce os niveis de proteínas tóxicas [130], [131], [132], [133]. Aínda que aínda non se observaron vínculos directos entre a inhibición de GSK-3 e a regulación transcricional de NRF2 de xenes que promoven a proteóstase, é razoable especular que a regulación descendente da actividade de GSK-3 daría lugar a un aumento dos niveis de NRF2, o que finalmente dará lugar a reforzos. proteóstase.
A actividade transcricional de NRF2, así como a capacidade celular para manter a proteóstase diminúen coa idade, o principal factor de risco para o desenvolvemento de enfermidades neurodexenerativas. É razoable pensar que o reforzo de NRF2 e, en consecuencia, a proteóstase, atrasaría, polo menos, a acumulación de agregados de proteínas e a neurodexeneración. De feito, o tratamento de fibroblastos senescentes humanos con ácido 18-glicirretínico (18--GA) triterpenoide promoveu a activación NRF2, o que levou á indución do proteasoma e á vida útil mellorada. Este estudo suxire que a activación farmacolóxica de NRF2 é posible incluso a finais da vida [86]. Ademais, un estudo posterior mostrou que este composto mediaba a activación do SKN-1 e do proteasoma en C.elegans con efectos beneficiosos sobre a progresión do AD en modelos de nematodos relevantes [134].
Todas as cousas consideradas, a inducción mediada por NRF2 de xenes relacionados coa proteostase parece ser beneficiosa en diferentes proteopatías.
Sulforaphane e os seus efectos sobre o cancro, a mortalidade, o envellecemento, o cerebro e o comportamento, as enfermidades cardíacas e moito máis
Os isotiocianatos son algúns dos compostos vexetais máis importantes que pode obter na súa dieta. Neste video fago o caso máis completo para eles que se fixo. ¿A atención curta? Saltar ao teu tema favorito premendo un dos puntos de tempo a continuación. Cadro de cronograma completo a continuación.
Seccións clave:
00: 01: 14 - Cáncer e mortalidade
00: 19: 04 - Envellecemento
00: 26: 30 - Cerebro e comportamento
00: 38: 06 - Recapitalización final
00: 40: 27 - Dose
Cadro de tempo completo:
00: 00: 34 - Introdución de sulforaphane, un foco principal do vídeo.
00: 01: 14 - Consumo e redución de vexetais cruciferos na mortalidade por todas as causas.
00: 02: 12 - Risco de cancro de próstata.
00: 02: 23 - Risco de cancro de vejiga.
00: 02: 34 - Cáncer de pulmón en risco de fumadores.
00: 02: 48 - Risco de cancro de mama.
00: 03: 13 - hipotético: e se xa ten cancro? (intervencionista)
00: 03: 35 - Mecanismo plausible que conduce os datos asociativos de cancro e mortalidade.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Evidencia animal que mostra un forte efecto do extracto de brotes de brócoli no desenvolvemento de tumores vesicales en ratas.
00: 06: 06 - Efecto da suplementación directa de sulforaphane en pacientes con cancro de próstata.
00: 07: 09 - Bioacumulación de metabolitos de isotiocianato no tecido de mama real.
00: 08: 32 - Inhibición das células nais do cancro de mama.
00: 08: 53 - Lección de historia: as brassicas establecéronse con propiedades de saúde mesmo na Roma antiga.
00: 09: 16 - A capacidade de Sulforaphane para mellorar a excreción de carcinóxenos (benceno, acroleína).
00: 09: 51 - NRF2 como un cambio xenético a través de elementos de resposta antioxidante.
00: 10: 10 - Como a activación de NRF2 aumenta a excreción de carcinóxenos a través de glutatión-S-conjugados.
00: 10: 34 - As coles de Bruxelas aumentan a glutatión-S-transferasa e reducen o dano do ADN.
00: 11: 20 - A bebida de brote de brócoli aumenta a excreción de benceno por 61%.
00: 13: 31 - O homogeneio de brotes de brócoli aumenta as encimas antioxidantes nas vías aéreas superiores.
00: 15: 45 - Consumo de vexetais cruciferos e mortalidade cardíaca.
00: 16: 55 - O polbo de brócolis mellora os lípidos sanguíneos eo risco de enfermidade cardíaca en diabéticos tipo 2.
00: 19: 04 - Comezo da sección de envellecemento.
00: 19: 21 - A dieta enriquecida con Sulforaphane mellora a vida útil dos escaravellos de 15 a 30% (en certas condicións).
00: 20: 34 - Importancia da baixa inflamación por lonxevidade.
00: 22: 05 - As verduras cruciferas e os brotes de brócoli parecen reducir unha gran variedade de marcadores inflamatorios en humanos.
00: 23: 40 - Recapitalización media: cancro, seccións de envellecemento
00: 24: 14 - Os estudos do rato suxiren que o sulforaphano pode mellorar a función inmune adaptativa na vellez.
00: 25: 18 - Sulforaphane mellorou o crecemento do cabelo nun modelo de calvície de rato. Imaxe en 00: 26: 10.
00: 26: 30 - Comezo da sección do cerebro e do comportamento.
00: 27: 18 - Efecto do extracto de brotes de brócoli no autismo.
00: 27: 48 - Efecto da glucoraphanina na esquizofrenia.
00: 28: 17 - Inicio da discusión de depresión (mecanismo plausible e estudos).
00: 31: 21 - O estudo do rato usando 10 diferentes modelos de depresión inducida polo estrés mostran sulforaphane igualmente efectivo como a fluoxetina (prozac).
00: 32: 00 - O estudo mostra que a inxestión directa de glucoraphanina en ratos é igualmente eficaz na prevención da depresión do modelo de estrés da derrota social.
00: 33: 01 - Inicio da sección de neurodegeneración.
00: 33: 30 - Sulforaphane e enfermidade de Alzheimer.
00: 33: 44 - Sulforaphane e enfermidade de Parkinson.
00: 33: 51 - Sulforaphane e enfermidade de Hungtington.
00: 34: 13 - Sulforaphane aumenta as proteínas de choque térmico.
00: 34: 43 - Inicio da sección traumática de lesións cerebrais.
00: 35: 01 - Sulforaphane inxectado inmediatamente despois de que o TBI mellore a memoria (estudo do rato).
00: 35: 55 - Sulforaphane e plasticidade neuronal.
00: 36: 32 - Sulforaphane mellora a aprendizaxe en modelo de diabetes tipo II en ratos.
00: 37: 19 - Distrofia muscular sulforaphana e duxena.
00: 37: 44 - Inhibición da myostatina nas células satélite do músculo (in vitro).
00: 38: 06 - Recapitulación de última hora: mortalidade e cancro, danos no ADN, estrés oxidativo e inflamación, excreción de benceno, enfermidade cardiovascular, diabetes tipo II, efectos sobre o cerebro (depresión, autismo, esquizofrenia e neurodegeneración), vía NRF2.
00: 40: 27 - Pensamentos en descubrir unha dose de brotes de brócoli ou sulforaphane.
00: 41: 01 - Anécdotas sobre o xermelo na casa.
00: 43: 14 - Sobre as temperaturas de cocción e actividade sulforaphane.
00: 43: 45 - Conversión de bacterias gut de sulforaphane a partir de glucoraphanin.
00: 44: 24 - Os suplementos funcionan mellor cando se combinan con mirosinasa activa de vexetais.
00: 44: 56 - Técnicas de cociña e vexetais crucíferos.
00: 46: 06 - Isotiocianatos como goitrógenos.
O factor relacionado co factor nuclear 2 (NF-E2) derivado da eritroide, tamén coñecido como Nrf2, é un factor de transcrición que regula a expresión dunha variedade de enzimas antioxidantes e desintoxicantes. Os estudos de investigación tamén demostraron o seu papel no control do estrés oxidativo. A maioría das enfermidades neurodexenerativas, como a enfermidade de Alzheimer e a enfermidade de Parkinson, caracterízanse por estrés oxidativo e inflamación crónica, obxectivos comúns de O tratamento con Nrf2 achégase. Dr. Alex Jimenez DC, CCST Insight
Observacións finais
Factor de transcrición NRF2 orquestra unha resposta proteostática detectando e modulando cambios na UPR, UPS e autofagia (Fig. 4). En consecuencia, demostrouse que a falta de NRF2 agrava a proteopopatía, o que suxire que NRF2 é necesario para un aclaramento óptimo das proteínas. En conxunto, podemos especular que NRF2 pode ser un obxectivo terapéutico interesante para as proteopatopatías.
Figura 4 NRF2 como un núcleo que conecta sinais de emerxencia derivadas de proteotóxicos para unha resposta de transcrición protectora. A acumulación de proteínas despregadas / mal páxinas levará á activación da resposta proteica despregada (EPU) no ER. A activación de PERK ou MAPK pode resultar na inducción transcricional do Gpx8 residente en ER e de varias enzimas que regulan os niveis de GSH, críticos para asegurar a correcta dobraxe das proteínas. Os agregados de proteínas inhiben a actividade do proteasoma (UPS), probablemente evitando a degradación de NRF2. NRF2 demostrou que modula específicamente a transcrición dos xenes Psma3, Psma6, Psmb1, Psmb5 e Pomp. Outras subunidades foron reguladas de xeito dependente de NRF2 en resposta a D3T, probablemente ampliando a lista de subunidades de proteasomas reguladas por NRF2. A autofagia é a principal vía para a degradación dos agregados de proteínas. A autofaxia tamén regula NRF2, conectando esta vía de degradación coa inducción transcricional de NRF2 de p62, Ndp52, Ulk1, Atg2b, Atg4c, Atg5, Atg7 e Gabarapl1.
Segundo o artigo anterior, aínda que os síntomas das enfermidades neurodexenerativas poden tratarse a través dunha variedade de opcións de tratamento, os estudos demostraron que a activación de Nrf2 pode ser un enfoque de tratamento útil. Porque Os activadores de Nrf2 apuntan a grandes mecanismos da enfermidade, todas as enfermidades neurodexenerativas poden beneficiarse do uso do factor de transcrición Nrf2. Os achados de Nrf2 revolucionaron o tratamento de enfermidades neurodexenerativas. O alcance da nosa información limítase a problemas de saúde quiropráctica e espiñal. Para falar do asunto, non dubide en preguntarlle ao doutor Jiménez ou contactar connosco en 915-850-0900 .
Discusión temática adicional: aliviar a dor no xeonllo sen cirurxía
A dor no xeonllo é un síntoma ben coñecido que pode ocorrer debido a unha variedade de lesións e / ou afeccións no xeonllo, incluíndo lesións deportivas. O xeonllo é unha das articulacións máis complexas do corpo humano xa que está composto pola intersección de catro ósos, catro ligamentos, varios tendóns, dous meniscos e cartilaxe. Segundo a Academia Americana de Médicos de Familia, as causas máis comúns de dor no xeonllo inclúen a subluxación rotular, a tendinite rotuliana ou o xeonllo do saltador e a enfermidade de Osgood-Schlatter. Aínda que a dor no xeonllo é máis probable que se produza en persoas maiores de 60 anos, a dor no xeonllo tamén pode ocorrer en nenos e adolescentes. A dor no xeonllo pódese tratar na casa seguindo os métodos RICE, con todo, as lesións graves no xeonllo poden requirir atención médica inmediata, incluída a atención quiropráctica.
O estrés oxidativo descríbese como dano celular causado por radicais libres ou moléculas inestables, o que pode afectar ao final a función sa. O corpo humano crea radicais libres para neutralizar as bacterias e os virus, pero os factores externos, como o osíxeno, a contaminación ea radiación, moitas veces producen tamén radicais libres. O estrés oxidativo asociouse a numerosos problemas de saúde.
O estrés oxidativo e outros factores estresantes activan mecanismos de protección internos que poden axudar a regular a resposta antioxidante do corpo humano. Nrf2 é unha proteína que detecta os niveis de estrés oxidativo e permite ás células protexerse dos factores internos e externos. Tamén se demostrou que Nrf2 axudou a regular os xenes implicados na produción de encimas antioxidantes e xenes de estrés-resposta. O obxectivo do artigo seguinte é explicar o efectos de Nrf2 en cancro.
Abstracto
A vía Keap1-Nrf2 é o principal regulador das respostas citoprotectores ao estrés oxidativo e electrofílico. Aínda que as vías de sinalización celular provocadas polo factor de transcrición Nrf2 prevén o inicio e progresión do cancro en tecidos normais e premalignas, en células totalmente malignas, a actividade Nrf2 proporciona unha vantaxe de crecemento aumentando a quimioterapia do cancro e potenciando o crecemento das células tumorais. Nesta revisión gráfica, proporcionamos unha visión xeral da ruta Keap1-Nrf2 ea súa disregulación nas células cancerosas. Tamén resumimos brevemente as consecuencias da activación constitutiva de Nrf2 nas células cancerosas e como isto pode ser explotado na terapia xenética do cancro.
Palabras clave:Nrf2, Keap1, Cáncer, elemento de resposta antioxidante, terapia xenética
introdución
A vía Keap1-Nrf2 é o principal regulador das respostas citoprotectoras ás tensións endóxenas e esóxenas causadas por especies reactivas de osíxeno (ROS) e electrófilos [1]. As proteínas clave de sinalización dentro da vía son o factor de transcrición Nrf2 (factor 2 relacionado co factor eritroide 2) que se une xunto con pequenas proteínas Maf ao elemento de resposta antioxidante (ARE) nas rexións reguladoras dos xenes diana e Keap1 (Kelch ECH asociando a proteína 1), unha proteína represora que se une a Nrf2 e promove a súa degradación pola vía do proteasoma da ubiquitina (Fig. 1). Keap1 é unha proteína moi rica en cisteína, o ratón Keap1 ten un total de 25 e 27 residuos de cisteína humana, a maioría dos cales poden ser modificados in vitro por diferentes oxidantes e electrófilos [2]. Demostrouse que tres destes residuos, C151, C273 e C288, xogan un papel funcional alterando a conformación de Keap1 que leva á translocación nuclear de Nrf2 e posterior expresión xénica diana [3] (Fig. 1). Non se coñece o mecanismo exacto polo cal as modificacións da cisteína en Keap1 conducen á activación de Nrf2, pero os dous modelos predominantes pero non excluíntes mutuamente son (1) o modelo hinghing e latch , no que as modificacións de Keap1 nos residuos de tiol que residen no IVR de Keap1 interrompa a interacción con Nrf2 provocando unha desalineamiento dos residuos de lisina dentro de Nrf2 que xa non se poden poliubiquitinilar e (2) o modelo no que a modificación do tiol provoca a disociación de Cul3 de Keap1 [3]. En ambos modelos, o Keap2 modificado por indutor e unido a Nrf1 está inactivado e, en consecuencia, as proteínas Nrf2 recentemente sintetizadas obvian Keap1 e traslácanse ao núcleo, únense ao ARE e conducen a expresión de xenes obxectivo Nrf2 como NAD (P) H quinona oxidorreductase 1 (NQO1), hemoxixenase 1 (HMOX1), glutamato-cisteína ligase (GCL) e glutatión S transferases (GST) (Fig. 2). Ademais das modificacións dos tioles de Keap1 que resultan na indución dos xenes obxectivo Nrf2, proteínas como p21 e p62 poden unirse a Nrf2 ou Keap1 perturbando así a interacción entre Nrf2 e Keap1 [1], [3] (Fig. 3).
Fig. 1. Estruturas de Nrf2 e Keap1 e o código de cisteína. (A) Nrf2 consta de 589 aminoácidos e ten seis dominios evolutivamente moi conservados, Neh1-6. Neh1 contén un motivo bZip, unha estrutura básica da cremalleira leucina (L-Zip), onde a rexión básica é responsable do recoñecemento do ADN e a L-Zip media a dimerización con pequenas proteínas Maf. Neh6 funciona como un degrón para mediar a degradación de Nrf2 no núcleo. Neh4 e 5 son dominios de transactivación. Neh2 contén motivos ETGE e DLG, que son necesarios para a interacción con Keap1, e unha rexión hidrófila de residuos de lisina (7 K), indispensables para a poliubiquitinación dependente de Keap1 e a degradación de Nrf2. (B) Keap1 consta de 624 residuos de aminoácidos e ten cinco dominios. Os dous motivos de interacción proteínica, o dominio BTB e o dominio Kelch, están separados pola rexión intermedia (IVR). O dominio BTB xunto coa porción N-terminal do IVR media a homodimerización de Keap1 e a unión con Cullin3 (Cul3). O dominio Kelch e a rexión do terminal C median a interacción con Neh2. (C) Nrf2 interactúa con dúas moléculas de Keap1 a través dos seus motivos Neh2 ETGE e DLG. Tanto ETGE como DLG únense a sitios similares na superficie inferior do motivo Keap1 Kelch. (D) Keap1 é rico en residuos de cisteína, con 27 cisteínas en proteína humana. Algunhas destas cisteínas localízanse preto de residuos básicos e, polo tanto, son excelentes obxectivos de electrófilos e oxidantes. O patrón de modificación dos residuos de cisteína por electrófilos coñécese como código de cisteína. A hipótese do código da cisteína propón que axentes activadores de Nrf2 estruturalmente diferentes afectan ás diferentes cisteínas de Keap1. As modificacións da cisteína levan a cambios conformacionais no Keap1 que interrompe a interacción entre os dominios Nrf2 DLG e Keap1 Kelch, inhibindo así a poliubiquitinación de Nrf2. Demostrouse a importancia funcional de Cys151, Cys273 e Cys288, xa que Cys273 e Cys288 son necesarios para a supresión de Nrf2 e Cys151 para a activación de Nrf2 por indutores [1], [3].
Fig. 2. Ruta de sinalización Nrf2-Keap1. (A e B) en condicións basales, dúas moléculas de Keap1 únense a Nrf2 e Nrf2 está poliubicada polo complexo de ligase E3 baseado en Cul3. Esta poliubiquitilación produce unha degradación rápida de Nrf2 polo proteasoma. Unha pequena proporción de Nrf2 escapa ao complexo inhibidor e acumúlase no núcleo para mediar a expresión xénica basal ARE, mantendo así a homeostase celular. (C) En condicións de estrés, os inductores modifican as cisteínas Keap1 que levan á inhibición da ubicuidade de Nrf2 mediante a disociación do complexo inhibidor. (D) Segundo o modelo de bisagra e bloqueo, a modificación dos residuos específicos de cisteína de Keap1 conduce a cambios conformacionais en Keap1, obtendo o desprendemento do motivo Nrf2 DLG de Keap1. A ubiquitinación de Nrf2 é interrompida, pero mantense a unión co motivo ETGE. (E) No modelo de disociación Keap1-Cul3, a unión de Keap1 e Cul3 é interrompida en resposta aos electrófilos, levando á fuga de Nrf2 desde o sistema de ubiquitinación. En ambos os modelos suxeridos, o Keap2 ligado a Nrf1 e modificado polo inductor está inactivo e, consecuentemente, as proteínas Nrf2 recén sintetizadas ignoran Keap1 e se translocan ao núcleo, únense ao Elemento de resposta antioxidante (ARE) e dirixe a expresión do obxectivo Nrf2 xenes como NQO1, HMOX1, GCL e GST [1], [3].
Fig. 3. Mecanismos para a acumulación nuclear constitutiva de Nrf2 en cancro. (A) As mutacións somáticas en Nrf2 ou Keap1 interrompen a interacción destas dúas proteínas. En Nrf2, as mutacións afectan os motivos ETGE e DLG, pero nas mutacións Keap1 hai unha distribución máis uniforme. Ademais, a activación do oncogênio, como o KrasG12D [5] ou a interrupción dos supresores tumorales, como PTEN [11] pode levar á indución transcripcional de Nrf2 e un aumento no Nrf2 nuclear. (B) A hipermetilación do promotor Keap1 no cancro de próstata e pulmón orixina unha redución da expresión mRNA de Keap1, que aumenta a acumulación nuclear de Nrf2 [6], [7]. (C) No carcinoma renal papilar familiar, a perda da actividade de enzima de hidratasa de fumarato conduce á acumulación de fumarato e posteriormente á succinación dos residuos de cisteína Keap1 (2SC). Esta modificación postraducional leva á interrupción da interacción Keap1-Nrf2 e acumulación nuclear de Nrf2 [8], [9]. (D) A acumulación de proteínas disruptoras como p62 e p21 pode perturbar o enlace Nrf2-Keap1 e producir un aumento no Nrf2 nuclear. p62 únese a KeapXUMX que se solapa co peto de conexión para Nrf1 e p2 interactúa directamente cos motivos DLG e ETGE de Nrf21, competindo así con Keap2 [1].
Mecanismos de activación e desregulación de Nrf2 en cancro
Aínda que a citoprotección proporcionada pola activación Nrf2 é importante para a quimioprevención do cancro en tecidos normais e premalignos, en células totalmente malignas. A actividade Nrf2 proporciona unha vantaxe de crecemento aumentando a quimiorresistencia do cancro e mellorando o crecemento das células tumorais [4]. Varios mecanismos polos cales a vía de sinalización Nrf2 está constitutivamente activada en varios tipos de cancro foron descritos: (1) mutacións somáticas en Keap1 ou o dominio de unión Keap1 de Nrf2 que interrompe a súa interacción; (2) silenciamento epigenético da expresión Keap1 que leva a unha represión defectuosa de Nrf2; (3) acumulación de proteínas disruptoras como p62 que levan á disociación do complexo Keap1-Nrf2; (4) indución transcripcional de Nrf2 por oncogénico K-Ras, B-Raf e c-Myc; (5), a modificación postraducional das cisteínas Keap1 por succinilación que ocorre no carcinoma renal papilar familiar debido á perda da actividade de encimas de hidratasa de fumarato [3], [4], [5], [6], [7], [ 8], [9], [10] (Fig. 3). A abundante proteína Nrf2 constitúe unha maior expresión de xenes implicados no metabolismo do fármaco aumentando así a resistencia aos medicamentos quimioterapéuticos e á radioterapia. Ademais, o alto nivel de proteína Nrf2 está asociado cun mal pronóstico de cancro [4]. O Nrf2 sobreactivo tamén afecta a proliferación celular dirixindo a glicosa ea glutamina cara ás vías anabólicas que aumentan a síntese de purinas e inflúen na vía de fosfato de pentosa para promover a proliferación celular [11] (Fig. 4).
Fig. 4. O papel dobre de Nrf2 na tumorigénesis. Baixo condicións fisiolóxicas, os baixos niveis de Nrf2 nuclear son suficientes para o mantemento da homeostase celular. Nrf2 inhibe a iniciación do tumor ea metástasis do cancro eliminando carcinóxenos, ROS e outros axentes danados polo ADN. Durante a tumorigénesis, a acumulación de dano no ADN leva á hiperactividade constitutiva de Nrf2 que axuda ás células malignas autónomas a sufrir altos niveis de ROS endóxeno e para evitar a apoptose. Os niveis Nrf2 elevados de forma persistente activan xenes metabólicos ademais dos xenes citoprotectores que contribúen á reprogramación metabólica e á proliferación celular. Os cancros con niveis elevados de Nrf2 están asociados a un mal pronóstico por radio e quimiorresistencia e proliferación de células agresivas de cancro. Así, a actividade da vía Nrf2 é protectora nas etapas iniciais da tumorigénesis, pero é prexudicial nas etapas posteriores. Polo tanto, para a prevención do cancro, o aumento da actividade Nrf2 segue sendo un enfoque importante mentres que para o tratamento do cancro, a inhibición de Nrf2 é desexable [4], [11].
Tendo en conta que a alta actividade de Nrf2 ocorre normalmente en células cancerosas con resultados adversos, hai unha necesidade de que as terapias inhiban a Nrf2. Desafortunadamente, debido a similitudes estruturais con algúns outros membros da familia bZip, o desenvolvemento de inhibidores Nrf2 específicos é unha tarefa desafiante e só se publicaron algúns estudos da inhibición de Nrf2 ata a data. Mediante a selección de produtos naturais, Ren et al. [12] identificou un composto antineoplástico brusatol como un inhibidor de Nrf2 que mellora a eficacia quimioterapéutica do cisplatino. Ademais, inhibidores de PI3K [11], [13] e Nrf2 siRNA [14] foron usados para inhibir Nrf2 nas células cancerosas. Recentemente utilizamos un enfoque alternativo, coñecido como terapia xénica de suicidio do cancro, para dirixir células cancerosas con altos niveis de Nrf2. Os vectores lentivirales con [2] que conteñen Nrf15 que conteñen timidina quinasa (TK) transfírense a células cancerosas con actividade ARE elevada e as células son tratadas cun pro-fármaco, ganciclovir (GCV). O GCV metabolízase a GCV-monofosfato, que é fosforilado aínda máis por quinasas móbiles nunha forma de trifosfato tóxico [16] (Fig. 5). Isto leva a unha matanza efectiva de células tumorais que non conteñen TK, pero tamén as células veciñas debido ao efecto de portador [17]. A terapia xenética regulada por TK / GCV pode mellorarse mediante a combinación dun doxorubicina do cancro do axente quimioterapéutico co tratamento [16], apoiando a noción de que este enfoque podería ser útil en combinación coas terapias tradicionais.
Fig. 5. Terapia xénica suicida. A acumulación nuclear Nrf2 constitucional nas células cancerosas pode ser explotada usando o vector viral impulsado por Nrf2 para a terapia xénica de suicidio por cancro [16]. Neste enfoque, o vector lentiviral (LV) que expresa a timidina quinasa (TK) baixo un promotor SV40 mínimo con catro AREs transdúcese a células de adenocarcinoma pulmonar. Os niveis elevados de Nrf2 nuclear levan a unha forte expresión de conexión TK a través de Nrf2. As células son entón tratadas cun pro-fármaco, ganciclovir (GCV), que é fosforilado por TK. O GCV trifosforilado interrompeu a síntese de ADN e conduce a unha matanza efectiva de células tumorais que non conteñen TK, pero tamén as células veciñas debido ao efecto do espectador.
Nrf2 é un regulador mestre que activa a produción de poderosos antioxidantes no corpo humano que axuda a eliminar o estrés oxidativo. Varias enzimas antioxidantes, como a superóxido dismutasa ou SOD, glutatión e catalase, tamén se activan a través da vía Nrf2. Ademais, certos fitoquímicos como cúrcuma, ashwagandha, bacopa, té verde e cardo de leite activan Nrf2. Estudos de investigación descubriron iso Activación de Nrf2 pode mellorar naturalmente a protección celular e restaurar o equilibrio para o corpo humano.
Dr. Alex Jimenez DC, CCST Insight
Sulforaphane e os seus efectos sobre o cancro, a mortalidade, o envellecemento, o cerebro e o comportamento, as enfermidades cardíacas e moito máis
Os isotiocianatos son algúns dos compostos vexetais máis importantes que pode obter na súa dieta. Neste video fago o caso máis completo para eles que se fixo. ¿A atención curta? Saltar ao teu tema favorito premendo un dos puntos de tempo a continuación. Cadro de cronograma completo a continuación.
Seccións clave:
00: 01: 14 - Cáncer e mortalidade
00: 19: 04 - Envellecemento
00: 26: 30 - Cerebro e comportamento
00: 38: 06 - Recapitalización final
00: 40: 27 - Dose
Cadro de tempo completo:
00: 00: 34 - Introdución de sulforaphane, un foco principal do vídeo.
00: 01: 14 - Consumo e redución de vexetais cruciferos na mortalidade por todas as causas.
00: 02: 12 - Risco de cancro de próstata.
00: 02: 23 - Risco de cancro de vejiga.
00: 02: 34 - Cáncer de pulmón en risco de fumadores.
00: 02: 48 - Risco de cancro de mama.
00: 03: 13 - hipotético: e se xa ten cancro? (intervencionista)
00: 03: 35 - Mecanismo plausible que conduce os datos asociativos de cancro e mortalidade.
00: 04: 38 - Sulforaphane e cancro.
00: 05: 32 - Evidencia animal que mostra un forte efecto do extracto de brotes de brócoli no desenvolvemento de tumores vesicales en ratas.
00: 06: 06 - Efecto da suplementación directa de sulforaphane en pacientes con cancro de próstata.
00: 07: 09 - Bioacumulación de metabolitos de isotiocianato no tecido de mama real.
00: 08: 32 - Inhibición das células nais do cancro de mama.
00: 08: 53 - Lección de historia: as brassicas establecéronse con propiedades de saúde mesmo na Roma antiga.
00: 09: 16 - A capacidade de Sulforaphane para mellorar a excreción de carcinóxenos (benceno, acroleína).
00: 09: 51 - NRF2 como un cambio xenético a través de elementos de resposta antioxidante.
00: 10: 10 - Como a activación de NRF2 aumenta a excreción de carcinóxenos a través de glutatión-S-conjugados.
00: 10: 34 - As coles de Bruxelas aumentan a glutatión-S-transferasa e reducen o dano do ADN.
00: 11: 20 - A bebida de brote de brócoli aumenta a excreción de benceno por 61%.
00: 13: 31 - O homogeneio de brotes de brócoli aumenta as encimas antioxidantes nas vías aéreas superiores.
00: 15: 45 - Consumo de vexetais cruciferos e mortalidade cardíaca.
00: 16: 55 - O polbo de brócolis mellora os lípidos sanguíneos eo risco de enfermidade cardíaca en diabéticos tipo 2.
00: 19: 04 - Comezo da sección de envellecemento.
00: 19: 21 - A dieta enriquecida con Sulforaphane mellora a vida útil dos escaravellos de 15 a 30% (en certas condicións).
00: 20: 34 - Importancia da baixa inflamación por lonxevidade.
00: 22: 05 - As verduras cruciferas e os brotes de brócoli parecen reducir unha gran variedade de marcadores inflamatorios en humanos.
00: 23: 40 - Recapitalización media: cancro, seccións de envellecemento
00: 24: 14 - Os estudos do rato suxiren que o sulforaphano pode mellorar a función inmune adaptativa na vellez.
00: 25: 18 - Sulforaphane mellorou o crecemento do cabelo nun modelo de calvície de rato. Imaxe en 00: 26: 10.
00: 26: 30 - Comezo da sección do cerebro e do comportamento.
00: 27: 18 - Efecto do extracto de brotes de brócoli no autismo.
00: 27: 48 - Efecto da glucoraphanina na esquizofrenia.
00: 28: 17 - Inicio da discusión de depresión (mecanismo plausible e estudos).
00: 31: 21 - O estudo do rato usando 10 diferentes modelos de depresión inducida polo estrés mostran sulforaphane igualmente efectivo como a fluoxetina (prozac).
00: 32: 00 - O estudo mostra que a inxestión directa de glucoraphanina en ratos é igualmente eficaz na prevención da depresión do modelo de estrés da derrota social.
00: 33: 01 - Inicio da sección de neurodegeneración.
00: 33: 30 - Sulforaphane e enfermidade de Alzheimer.
00: 33: 44 - Sulforaphane e enfermidade de Parkinson.
00: 33: 51 - Sulforaphane e enfermidade de Hungtington.
00: 34: 13 - Sulforaphane aumenta as proteínas de choque térmico.
00: 34: 43 - Inicio da sección traumática de lesións cerebrais.
00: 35: 01 - Sulforaphane inxectado inmediatamente despois de que o TBI mellore a memoria (estudo do rato).
00: 35: 55 - Sulforaphane e plasticidade neuronal.
00: 36: 32 - Sulforaphane mellora a aprendizaxe en modelo de diabetes tipo II en ratos.
00: 37: 19 - Distrofia muscular sulforaphana e duxena.
00: 37: 44 - Inhibición da myostatina nas células satélite do músculo (in vitro).
00: 38: 06 - Recapitulación de última hora: mortalidade e cancro, danos no ADN, estrés oxidativo e inflamación, excreción de benceno, enfermidade cardiovascular, diabetes tipo II, efectos sobre o cerebro (depresión, autismo, esquizofrenia e neurodegeneración), vía NRF2.
00: 40: 27 - Pensamentos en descubrir unha dose de brotes de brócoli ou sulforaphane.
00: 41: 01 - Anécdotas sobre o xermelo na casa.
00: 43: 14 - Sobre as temperaturas de cocción e actividade sulforaphane.
00: 43: 45 - Conversión de bacterias gut de sulforaphane a partir de glucoraphanin.
00: 44: 24 - Os suplementos funcionan mellor cando se combinan con mirosinasa activa de vexetais.
00: 44: 56 - Técnicas de cociña e vexetais crucíferos.
00: 46: 06 - Isotiocianatos como goitrógenos.
Grazas
Este traballo foi apoiado pola Academia de Finlandia, a Fundación Sigrid Juselius e as Organizacións de Cancro de Finlandia.
En conclusión, o factor nuclear (como o 2 derivado do eritroide), tamén coñecido como NFE2L2 ou Nrf2, é unha proteína que aumenta a produción de antioxidantes que protexen o corpo humano contra o estrés oxidativo. Como se describiu anteriormente, a estimulación da vía Nrf2 están a ser estudos para o tratamento de enfermidades causadas por estrés oxidativo, incluído o cancro. O alcance da nosa información limítase a problemas de saúde quiropráctica e espiñal. Para falar do asunto, non dubide en preguntarlle ao doutor Jiménez ou contactar connosco en 915-850-0900 .
Discusión temática adicional: aliviar a dor no xeonllo sen cirurxía
A dor no xeonllo é un síntoma ben coñecido que pode ocorrer debido a unha variedade de lesións e / ou afeccións no xeonllo, incluíndo lesións deportivas. O xeonllo é unha das articulacións máis complexas do corpo humano xa que se compón da intersección de catro ósos, catro ligamentos, varios tendóns, dous menisci e cartilaxe. Segundo a Academia Americana de Médicos Familiares, as causas máis comúns na dor do xeonllo inclúen a subluxación patelar, a tendinite patelar ou o xeonllo do jumper e a enfermidade de Osgood-Schlatter. Aínda que a dor de xeonllos é máis probable que ocorra en persoas con máis de 60 anos de idade, a dor no xeonllo tamén pode ocorrer en nenos e adolescentes. A dor de xeonllos pódese tratar na casa seguindo os métodos RICE, con todo, as lesións graves no xeonllo poden requirir atención médica inmediata, incluído o coidado quiropráctico.
O ADN soporta aproximadamente xenes 20,000, cada un ten un programa para a creación dunha proteína ou enzima requirida para un estilo de vida saudable. Cada un destes patróns debe estar constantemente regulado por unha especie de "promotor" que xestiona exactamente a cantidade de cada sustancia e / ou química que se xera e en que condicións tamén se desenvolverán.
Ao conectarse a un tipo particular das áreas de promotores como un switch, coñecido como Elemento de resposta antioxidante ou ARE, o Factor Nrf2Apoia a velocidade de creación de centos de xenes distintos que permiten ás células sobrevivir en circunstancias estresantes. Estes xenes xeran entón unha selección de encimas antioxidantes que desenvolven unha rede de defensa neutralizando os oxidantes e limpando os subprodutos tóxicos que quedaron na súa produción, ademais de axudar a restaurar os danos que causaron.
¿Que é o estrés oxidativo?
Varios oxidantes como o radical superóxido ou O2- e o peróxido de hidróxeno ou H2O2 creáronse coa práctica de queimar as substancias e / ou produtos químicos que sustentan o corpo humano. O corpo humano posúe encimas antioxidantes que neutralizan e desintoxican os alimentos e bebidas reactivos que consumimos. O Nrf2 modula a súa produción para manter o equilibrio e subliña a demanda de todos estes encimas. Este equilibrio pode ser interrompido por un par de factores, incluída a idade.
A medida que envellecemos, o corpo humano crea menos Nrf2 e este delicado equilibrio pode comezar gradualmente a virar cara ao lado oxidativo, un estado denominado estrés oxidativo. A enfermidade tamén pode causar a sobreprodución de oxidantes. Infeccións, alerxias e trastornos autoinmunes poden desencadear as nosas células inmunes a crear oxidantes reactivos, como o O2-. , H2O2, OH e HOCl, onde as células saudables danan e responden con inflamación. As enfermidades asociadas ao envellecemento, incluíndo ataques cardíacos, ictus, cancro e afeccións neurodexenerativas como a enfermidade de Alzheimer, tamén aumentan o desenvolvemento de oxidantes, xerando estrés e unha resposta inflamatoria.
Que son os activadores Nrf2?
A proteína Nrf2, tamén chamada factor de transcrición debido á forma na que pode soportar e controlar as enzimas e os xenes, é o elemento secreto dunha secuencia de reaccións bioquímicas dentro da célula que reacciona ás modificacións no equilibrio cognitivo e no equilibrio oxidativo. Os elementos sensoriais desta ruta modifican e descargan Nrf2, o que o desencadea para que se estenda ao núcleo da célula cara ao ADN. O Nrf2 pode alternativamente activar ou desactivar os xenes e encimas que soporta para protexer a célula.
Afortunadamente, unha variedade de sustancias que son activadores Nrf2 desenvolven a través do consumo de certas plantas e extractos utilizados hai séculos nos remedios tradicionais chineses e nativos americanos. Estes fitoquímicos parecen ser igual de poderosos con menos efectos secundarios, como os produtos farmacéuticos que activan Nrf2 que se están a usar hoxe en día.
Factor nuclear eritroide Factor relacionado con 2, máis coñecido como Nrf2, é un factor de transcrición que protexe a célula regulando xenes, enzimas e respostas antioxidantes. Os factores de transcrición son un tipo de proteína que se unen ao ADN para promover a creación de sustancias e produtos químicos específicos, incluídos glutatión S-transferases ou GST. A activación de Nrf2 induce a produción de proteínas activas que presentan unha potente capacidade antioxidante que axuda a diminuír o estrés oxidativo.
Dr. Alex Jimenez DC, CCST Insight
A ciencia detrás da activación Nrf2
Unha vez que se creou o suplemento alimenticio Nrf2 inicial en 2004, coñécese información mínima sobre a función da ruta Nrf2. Aproximadamente os xornais 200 na literatura sobre Nrf2, tamén coñecido como factor nuclear como 2 ou NFE2L2, existían e os investigadores só comezaban a descubrir a resposta antioxidante de Nrf2 en mamíferos. A partir de 2017, porén, foron impresos os estudos académicos 9,300 sobre este "regulador principal".
En realidade, Nrf2 regula moitos enzimas antioxidantes que non se relacionan cos xenes, no seu lugar, ofrecen protección contra unha variedade de circunstancias relacionadas co estrés que se atopan por células, órganos e, en última instancia, por organismos, en condicións sanas e patolóxicas. Con base nesta nova cantidade de información de estudos académicos publicados, os investigadores poden agora desenvolverse mellor Suplemento dietético Nrf2.
A partir de 2007, os estudos de investigación demostraron a complexa función da vía Nrf2. Atopáronse activadores Nrf2 que imitan factores de diferentes estruturas dentro do corpo humano. A través destas vías, os activadores Nrf2 foron equipados para sentir as condicións cambiantes en toda a célula para manter o equilibrio e responder aos requirimentos evolutivos dos xenes.
Por que usar os suplementos que activan Nrf2?
Como as habilidades de activación Nrf2 diminúen coa idade en organismos, os cambios poden comezar a ocorrer. Os estudos de investigación demostraron que o foco de Nrf2 nas células declina coa idade, mostrando marcadores aumentados de estrés oxidativo. Unha variedade de enfermidades relacionadas coa idade como a arteriosclerose e enfermidades cardiovasculares, artrite, cancro, obesidade, diabetes tipo 2, hipertensión, cataratas e enfermidade de Alzheimer, así como as enfermidades de Parkinson poden desenvolverse debido a estes cambios. O estrés oxidativo atopouse con estes problemas de saúde.
Ao estimular a capacidade da célula para aumentar a produción de activadores Nrf2, Suplemento dietético Nrf2 pode axudar a revivir a capacidade do propio corpo humano de contrarrestar os efectos do estrés oxidativo. Os ácidos graxos poliinsaturados ou PUFA son unha das moléculas máis fácilmente oxidadas e son particularmente vulnerables a sufrir danos causados por radicais libres. O ácido tiobarbitúrico, ou a TBARS, a produción pode aumentar coa idade, o que indica un estrés oxidativo elevado xunto cunha caída nas vías reguladas por Nrf2.
Bioloxicamente, a indución xénica é un mecanismo moi lento, que xeralmente require horas para transferirse a través dunha vía. Como resultado, moitos encimas posúen os seus propios interruptores de encendido / apagado que poden ser desencadeados en minutos por diferentes encimas reguladores. Os investigadores desenvolveron composicións propietarias de activadores Nrf2 que utilizan esta base de coñecemento de activación. A activación de Nrf2 componse non só de que o factor de transcrición Nrf2 se descarga do seu inhibidor e migra ao núcleo celular, senón que tamén se une a secuencias específicas de ADN para fomentar a expresión xénica citoprotectora, regulando o ritmo no que Nrf2 é sacado do núcleo.
Comprender o procedemento de eliminación e a activación de Nrf2 no corpo humano permitiu aos investigadores construír combinacións de diferentes activadores de Nrf2 para lograr a reflexión dos xenes a través da súa modulación. A combinación da base de coñecemento, xunto coa gran variedade doutros estudos de investigación, axudaron a producir activadores Nrf2 para o seu uso como suplementos dietéticos. O alcance da nosa información limítase a problemas de saúde quiropráctica e espiñal. Para falar do asunto, non dubide en preguntarlle ao doutor Jiménez ou contactar connosco en 915-850-0900 .
Comisariado polo Dr. Alex Jiménez
Discusión temática adicional: aliviar a dor no xeonllo sen cirurxía
A dor no xeonllo é un síntoma ben coñecido que pode ocorrer debido a unha variedade de lesións e / ou afeccións no xeonllo, incluíndo lesións deportivas. O xeonllo é unha das articulacións máis complexas do corpo humano xa que se compón da intersección de catro ósos, catro ligamentos, varios tendóns, dous menisci e cartilaxe. Segundo a Academia Americana de Médicos Familiares, as causas máis comúns na dor do xeonllo inclúen a subluxación patelar, a tendinite patelar ou o xeonllo do jumper e a enfermidade de Osgood-Schlatter. Aínda que a dor de xeonllos é máis probable que ocorra en persoas con máis de 60 anos de idade, a dor no xeonllo tamén pode ocorrer en nenos e adolescentes. A dor de xeonllos pódese tratar na casa seguindo os métodos RICE, con todo, as lesións graves no xeonllo poden requirir atención médica inmediata, incluído o coidado quiropráctico.
Os antioxidantes son referidos científicamente como compostos que restrinxen o proceso de oxidación no corpo humano, que se non se controla, pode crear radicais libres que poidan desenvolver numerosas reaccións en cadea que poden causar danos celulares. Por fortuna, o corpo humano pode crear tales mecanismos inmunitarios incorporados; con todo, ao montar especies de osíxeno reactivo ou ROS, non se pode neutralizar, obsérvase unha pequena chama que se descarta cando se infunde con osíxeno, o dano está obrigado .
Para continuar expandiéndose sobre a metáfora da chama, o produto final de non ter a capacidade de neutralizar o impacto de ROS ou as especies reactivas de osíxeno é dano, así como inflamación, é dicir, o corpo humano está literalmente en chamas. O fantástico é que hai antioxidantes que poden axudar tremendamente a combater este problema de saúde e este antioxidante é glutatión. Aínda que se atopa en 1889, o efecto antioxidante de glutationa converteuse nun dos temas máis interesantes nos estudos de investigación modernos.
Mestre de antioxidantes: glutatión
A substancia poderosa é un tripéptido que se desenvolve a partir de cisteína, ácido glutámico e glicina. Debido á súa capacidade para protexer o corpo humano contra a creación de radicais libres, o glutatión pode axudar a promover un sistema inmunitario saudable. Baseado en Informes científicos en 2015, determinouse que a capacidade de glutatión para funcionar de xeito sinérgico con peroxirina e catalasa axuda a protexer as células contra o peróxido de hidróxeno. Esta fórmula sinérxica funciona contra especies reactivas de osíxeno, ou ROS. O glutatión, a peroxidredina ea catalase son elementos esenciais no aumento da homeostase celular, que é un proceso esencial de células saudables, tecidos e órganos completamente.
Ademais, o glutatión aumenta a estrutura e función do sistema inmunitario global utilizando o seu efecto importante sobre as funcións do linfocito. Segundo o Departamento de Inmunochemistry, adecuadamente complementando os niveis de glutationa no corpo humano pode mellorar enormemente as reaccións inmunolóxicas. A modo de exemplo, dous ensaios aleatorios controlados con placebo demostraron que o tratamento terapéutico dos pacientes con inmunodeficiencia cardíaca con N-acetil-cisteína ou NAC resultou, en ambos casos, nun crecemento substancial na maioría dos procesos inmunolóxicos que incluían un rexuvenecemento completo da actividade de células asasinas naturais. N-acetil-cisteína, ou NAC, usa o xofre de glutatión e combina-lo con moléculas velenosas, que logo se converten en solubles en auga e se descargan no corpo humano.
O glutatión tamén ten a capacidade de revitalizar o ácido lipoico e reciclar a vitamina C e E, que son necesarios para iniciar certos procesos do sistema mediante o envío de electróns para neutralizar os radicais libres. Baseado nun estudo de investigación de PLoS ONE, o glutatión afectou aos pacientes con diabetes metil, ou T2DM, e mycobacterium tuberculosis. Normalmente, os individuos con sistemas inmunes débiles tenden a mostrar maior exposición a M. tb, ou a mycobacterium tuberculosis, enfermidade ou infección. Ademais, as persoas con Type 2 diabetes mellitus ou T2DM son dúas ou tres veces máis propensos á tuberculose que as persoas sen T2DM. O estudo de investigación tamén suxeriu que aumentar os niveis de glutationa nos macrófagos illados dos pacientes con T2DM conduciu a un mellor control da enfermidade de M.Tb ou a infección. Estes resultados demostran que os niveis máis baixos de glutatión en pacientes con T2DM contribúen a unha maior oportunidade de enfermidade ou infección por M. tb. Ademais, depende Dietro Ghezzi en Brighton e Sussex Medical SchoolO estrés oxidativo pode causar unha estrutura e función do sistema inmune deficiente.
Afortunadamente, o glutatión desempeña un papel esencial no fortalecemento e control da inmunidade. A modo de exemplo, o glutatión é esencial para os procesos innatos e adaptativos dentro do sistema inmunitario, incluíndo a proliferación de linfocitos T, actividade fagocítica de neutrófilos polimorfonucleares e funcións de células dendríticas, que poden ser fundamentais porque estas están compostas por células que presentan antíxenos . A inmunidade meditada por células inclúe antígenos proteínicos que inicialmente empezan a degenerar nas vesículas endócitos dos macrófagos e as células dendríticas, polo tanto, os péptidos menores móstranse na superficie para activar a proliferación de células T específicas do antíxeno. Ademais, o glutatión axuda na creación de citocinas e é necesario manter a produción de interferón-gamma por células dendríticas, o que é importante para protexer contra patóxenos intracelulares, incluíndo micobacterias.
N-acetil-cisteína, ou NAC, científicamente referida como o precursor do glutatión, tamén é un antioxidante celular moi poderoso usado como antioxidante de esterilización de radicais libres. Recoñecido habitualmente polo seu papel en evitando a toxicidade do acetaminofenoDemostrouse que o NAC ou a N-acetil-cisteína posúe varios beneficios para a saúde e o benestar. Dacordo con Revista móbil, O NAC axuda a soportar unha resposta inflamatoria sa e pode afectar positivamente os traballos a longo prazo ou prematuro humano. O estudo de investigación concluíu que en mulleres con natalidade prematura previa e vaginosis bacteriana, 0.6 gramo de NAC por día tomado oralmente xunto coa progesterona despois da semana 16 do embarazo protexido contra a recurrencia de parto prematuro e mellora do resultado neonatal. En conclusión, os efectos positivos do NAC sobre a construción muscular tamén foron detectados. Despois de tres minutos de contraccións persistentes, houbo un aumento de potencia 15 por cento, demostrando como o NAC desempeña un papel fundamental na mellora da construción muscular e reduce a fatiga xeral durante o parto.
Os investigadores tamén descubriron que o NAC, ou a N-acetil-cisteína, pode beneficiar aos que teñen síndrome de ovario poliquístico ou SOP. O SOP, ou síndrome de ovario poliquístico, é unha enfermidade común relacionada coas glándulas endócrinas que afecta aproximadamente ao 5 ao 10 por cento das mulleres en idade reprodutiva. Nestes pacientes, existe un maior risco de experimentar síndrome metabólica, onde o uso de NAC axudou a restaurar os niveis e a sensibilidade saudables de insulina.
Insight do Dr. Alex Jimenez
O glutatión foi referido como o "mestre dos antioxidantes" debido ao seu papel fundamental na consecución e mantemento da saúde e benestar en xeral. Aínda que o corpo humano é capaz de producir o seu propio glutatión, a mala nutrición, a contaminación, as toxinas, o uso excesivo de medicamentos e / ou medicamentos, o estrés, o trauma, o envellecemento, a enfermidade ea radiación poden diminuír os niveis naturais de glutation. Isto pode facer que as persoas sexan máis susceptibles ao dano celular do estrés oxidativo, os radicais libres, as infeccións e o cancro. A suplementación de glutatión pode ter enormes beneficios no corpo humano. Xunto coas opcións alternativas de tratamento, como coidados quiroprácticos, os niveis de glutatión poden ser regulados de novo para mellorar o benestar.
Ademais, os profesionais sanitarios suxeriron implementar o uso de suplementos de glutatión xunto con outras opcións alternativas de tratamento, como atención quiropraxia, para mellorar aínda máis a saúde e o benestar en xeral. Os antioxidantes son importantes para manter o máximo de benestar, así como para inhibir a reacción en cadea de radicais libres que causan dano ou dano celular. Antioxidantes poderosos como o glutatión, como se mencionou anteriormente, finalmente axudan a regular o desenvolvemento destes radicais libres e proporcionan unha resposta máis saudable ao sistema inmunitario. Estudos de investigación descubriron iso atención quiropraxia Tamén pode desempeñar un papel esencial neste proceso, aumentando de forma natural a actividade dos antioxidantes no corpo humano. O coidado quiropráctico é un enfoque de tratamento seguro e eficaz que utiliza axustes espiñentos e manipulacións manuais para corrixir desalineamientos espiñais ou subluxacións para permitir que o corpo humano cura naturalmente sen o uso de medicamentos e / ou intervencións cirúrxicas.
Finalmente, os antioxidantes demostran as súas propiedades biolóxicas a través de moitos beneficios para a saúde, que poderían ser necesarios agora máis que nunca coa cada vez maior ataque de estrés, enfermidades e contaminación no noso mundo moderno, que contribúen a danar e / ou danar as células. . O glutatión e o seu precursor, o NAC ou a N-acetil-cisteína, seguen mostrando o seu poderoso estado no reino dos antioxidantes. Xunto con opcións de tratamento alternativas, como a atención quiropráctica, as persoas poden aproveitar todos os beneficios que este poderoso antioxidante ten para ofrecer. O alcance da nosa información limítase á quiropráctica, así como ás lesións e condicións da columna vertebral. Para falar do asunto, non dubide en preguntarlle ao doutor Jiménez ou contactar connosco en 915-850-0900 .
Comisariado polo Dr. Alex Jiménez
Temas adicionais: dor nas costas
Dor nas costas é unha das causas máis frecuentes de discapacidade e días perdidos no traballo en todo o mundo. En realidade, a dor lumbar atribúese como o segundo motivo máis frecuente para as consultas médicas, superadas en número por infeccións respiratorias superiores. Aproximadamente o 80 por cento da poboación experimentará algún tipo de dor nas costas polo menos unha vez ao longo da súa vida. A columna vertebral é unha estrutura complexa composta de ósos, articulacións, ligamentos e músculos, entre outros tecidos brandos. Por iso, feridas e / ou condicións agravadas, como discos herniados, pode levar a síntomas de dor nas costas. As lesións deportivas ou as lesións por accidentes automovilísticos adoitan ser a causa máis frecuente de dor nas costas, con todo, ás veces o movemento máis sinxelo pode ter resultados dolorosos. Afortunadamente, as opcións de tratamento alternativas, como o coidado quiropráctico, poden axudar a aliviar a dor nas costas mediante o uso de axustes espiñentos e manipulacións manuais, mellorando o alivio da dor.
Chiropractor baseado na ciencia Dr. Alexander Jimenez bota unha ollada estrés oxidativo, o que é, como afecta ao corpo e á defensa antioxidante para remediar a situación.
Esra Birben PhD, 1 Umit Murat Sahiner MD, 1 Cansin Sackesen MD, 1 Serpil Erzurum MD, 2 e Omer Kalayci, MD1
Resumo: As especies reactivas de osíxeno (ROS) son producidas por organismos vivos como resultado do metabolismo celular normal e de factores ambientais, como contaminantes do aire ou fume de cigarro. Os ROS son moléculas moi reactivas e poden danar estruturas celulares como hidratos de carbono, ácidos nucleicos, lípidos e proteínas e alterar as súas funcións. O cambio no equilibrio entre oxidantes e antioxidantes en favor dos oxidantes denomínase estrés oxidativo. A regulación do estado redutor e oxidante (redox) é fundamental para a viabilidade, activación, proliferación e función dos órganos celulares. Os organismos aeróbicos teñen sistemas antioxidantes integrados, que inclúen antioxidantes encimáticos e non encimáticos que adoitan ser eficaces para bloquear os efectos nocivos do ROS. Non obstante, en condicións patolóxicas, os sistemas antioxidantes poden verse desbordados. O estrés oxidativo contribúe a moitas enfermidades e enfermidades patolóxicas, incluíndo cancro, trastornos neurolóxicos, aterosclerose, hipertensión, isquemia / perfusión, diabetes, síndrome de dificultades respiratorias agudas, fibrosis pulmonar idiopática, enfermidade pulmonar obstructiva crónica e asma. Nesta revisión, resumimos os sistemas oxidantes e antioxidantes celulares e discutimos os efectos e mecanismos celulares do estrés oxidativo.
As especies reactivas do osíxeno (ROS) son producidas polos organismos vivos como resultado do metabolismo celular normal. A concentracións baixas a moderadas, funcionan nos procesos fisiolóxicos das células, pero a altas concentracións producen modificacións adversas dos compoñentes celulares, como lípidos, proteínas e ADN.1�6 O cambio de equilibrio entre oxidante/antioxidante a favor dos oxidantes. denomínase estrés oxidativo. O estrés oxidativo contribúe a moitas condicións patolóxicas, incluíndo cancro, trastornos neurolóxicos,7 aterosclerose, hipertensión, isquemia/perfusión,10 diabetes, síndrome de dificultad respiratoria aguda, fibrose pulmonar idiopática, enfermidade pulmonar obstrutiva crónica. ,11 e asma.14�15 Os organismos aeróbicos teñen sistemas antioxidantes integrados,� que inclúen antioxidantes enzimáticos e non enzimáticos que adoitan ser eficaces para bloquear os efectos nocivos das ROS. Non obstante, en condicións patolóxicas, os sistemas antioxidantes poden verse desbordados. Nesta revisión, resumimos os sistemas celulares oxidantes e antioxidantes e a regulación do estado redutor e oxidante (redox) nos estados de saúde e enfermidade.
OXIDANTES
Fontes endóxenas de ROS
Os ROS prodúcense a partir de osíxeno molecular como resultado do metabolismo celular normal. O ROS pode dividirse en grupos 2: radicais libres e non radicais. As moléculas que conteñen un ou máis electróns non aparelados e así dan unha reactividade á molécula son chamados de radicais libres. Cando os radicais libres 2 comparten os seus electróns non aparados, créanse formas non radicais. Os principais ROS de 3 que teñen significado fisiolóxico son o anión superóxido (O22), o radical hidroxilo (OH) eo peróxido de hidrogênio (H2O2). ROS resúmense na táboa 1.
O anión superóxido fórmase pola adición de 1 electrón ao osíxeno molecular.22 Este proceso está mediado pola nicotina adenina dinucleótido fosfato [NAD(P)H] oxidase ou a xantina oxidase ou polo sistema de transporte de electróns mitocondriais. O principal sitio para producir anión superóxido son as mitocondrias, a maquinaria da célula para producir adenosina trifosfato. Normalmente, os electróns transfírense a través da cadea de transporte de electróns mitocondriais para a redución do osíxeno a auga, pero aproximadamente do 1 ao 3% de todos os electróns escapan do sistema e producen superóxido. A NAD(P)H oxidase atópase en leucocitos polimorfonucleares, monocitos e macrófagos. Tras a fagocitose, estas células producen unha explosión de superóxido que conduce á actividade bactericida. O superóxido convértese en peróxido de hidróxeno pola acción das superóxido dismutases (SOD, EC 1.15.1.1). O peróxido de hidróxeno difunde facilmente pola membrana plasmática. O peróxido de hidróxeno tamén se produce pola xantina oxidase, aminoácido oxidase e NAD(P)H oxidase�23,24 e nos peroxisomas polo consumo de osíxeno molecular nas reaccións metabólicas. Nunha sucesión de reaccións chamadas reaccións de Haber-Weiss e Fenton, o H2O2 pode descompoñerse a OH2 en presenza de metais de transmisión como Fe21 ou Cu21.25.
Fe31 +�.O2�?Fe2 +�O2 Haber Weiss
Fe2 +�H2O2�?Fe3 +�OH�+ .OH Reacción de Fenton
O O 2 �por si mesmo pode reaccionar co H2 O2 e xerar OH�.26,27 O radical hidroxilo é o máis reactivo dos ROS e pode danar proteínas, lípidos e hidratos de carbono e ADN. Tamén pode iniciar a peroxidación lipídica tomando un electrón dos ácidos graxos poliinsaturados.
Os encimas granulocíticos expanden aínda máis a reactividade do H2O2 a través da peroxidasa eosinófila e a mieloperoxidase (MPO). Nos neutrófilos activados, o H2O2 é consumido pola MPO. En presenza de ión cloruro, o H2O2 convértese en ácido hipocloroso (HOCl). O HOCl é altamente oxidativo e desempeña un papel importante na matanza de axentes patóxenos nas vías respiratorias.28 Non obstante, o HOCl tamén pode reaccionar co ADN e inducir interaccións proteínicas co ADN e producir produtos de oxidación da pirimidina e engadir cloruro ás bases do ADN.29,30 Eosinophil peroxidase e MPO tamén contribúen ao estrés oxidativo por modificación de proteínas por haloxenacións, nitración e enlaces cruzados de proteínas a través de radicais tirosilo.
Outros radicais libres derivados de osíxeno son os radicales peroxilo (ROO $). A forma máis simple destes radicais é o radical hidroxióxido (HOO $) e ten un papel na peroxidación do ácido graxo. Os radicais libres poden disparar reaccións de cadea de peroxidación lipídica abstrayendo un átomo de hidróxeno dun carbono de metileno de cadea lateral. O radical lípido reacciona entón co osíxeno para producir radical peroxilo. O radical peróxilo inicia unha reacción en cadea e transforma os ácidos graxos poliinsaturados en hidroperóxidos lipídicos. Os hidroperóxidos lipídicos son moi inestables e fácilmente se descompoñen aos produtos secundarios, como aldehídos (como 4-hydroxy-2,3-nonenal) e malondialdehídos (MDA). Os isoprostanos son outro grupo de produtos de peroxidación de lípidos que se xeran a través da peroxidación do ácido araquidónico e tamén se atoparon elevados en plasma e condensados de alento de asmáticos. 34,35 A peroxidación dos lípidos perturba a integridade das membranas celulares e orixina a reestruturación da estrutura da membrana .
O peróxido de hidróxeno, o radical superóxido, o glutatión oxidado (GSSG), os MDAs, os isoprostanos, os carbonilos e a nitrotyrosina poden ser facilmente medidos a partir de mostras de lavado en plasma, sangue ou broncoalveolares como biomarcadores de oxidación mediante ensaios estandarizados.
Fonte exógena de oxidantes
Fume de cigarro
O fume de cigarro contén moitos oxidantes e radicais libres e compostos orgánicos, como superóxido e óxido nítrico. 36 Ademais, a inhalación do fume do cigarro no pulmón tamén activa algúns mecanismos endóxenos, como a acumulación de neutrófilos e macrófagos, que aumentan aínda máis a lesión oxidante .
Exposición de ozono
A exposición ao ozono pode causar peroxidación lipídica e inducir o fluxo de neutrófilos no epitelio das vías respiratorias. A exposición a ozono a curto prazo tamén causa a liberación de mediadores inflamatorios, como MPO, proteínas catiónicas eosinófilas e tamén lactato deshidroxenase e albúmina. 37 Aínda en individuos sans, a exposición ao ozono causa unha redución nas funcións pulmonares. 38 Cho et al39 demostraron que As partículas (mestura de partículas sólidas e gotitas de líquido suspendidas no aire) catalizan a redución do osíxeno.
Hiperoxia
A hiperoxia refírese a condicións de niveis máis elevados de osíxeno que a presión parcial normal do osíxeno nos pulmóns ou outros tecidos do corpo. Leva a unha maior produción de especies reactivas de osíxeno e nitróxeno. 40,41
Radiación ionizante
A radiación ionizante, en presenza de O2, converte os radicais hidroxilo, superóxido e radicais orgánicos en peróxido de hidróxeno e hidroperóxidos orgánicos. Estas especies de hidroperóxido reaccionan con ións metálicos activos redox, como Fe e Cu, mediante reaccións de Fenton e inducen así estrés oxidativo.42,43 Narayanan et al44 demostraron que os fibroblastos que estaban expostos a partículas alfa tiñan aumentos significativos en O2 2. e H2O2 intracelulares. produción a través da NADPH oxidasa unida á membrana plasmática.44 Moléculas de transducción de sinais, como a quinasa 1 e 2 regulada polo sinal extracelular (ERK1 / 2), a quinasa N-terminal c-xuño (JNK) e p38, e factores de transcrición, como actívase a proteína activadora-1 (AP-1), o factor nuclear-kB (NF-kB) e p53, que resultan na expresión de xenes relacionados coa resposta á radiación. 45-50 Os fotóns ultravioleta A (UVA) desencadean reaccións oxidativas por excitación de fotosensibilizadores endóxenos, como porfirinas, NADPH oxidasa e riboflavinas. A 8-Oxo-7,8- dihidroguanina (8-oxoGua) é o principal produto de oxidación do ADN mediado por UVA formado pola oxidación do radical OH, os oxidantes de 1 electrón e o osíxeno sinxelo que reacciona principalmente coa guanina.51 A formación da guanina demostrouse que o catión radical no ADN illado prodúcese de xeito eficiente a través do efecto directo da radiación ionizante.52,53 Despois da exposición a radiacións ionizantes, o nivel intracelular de glutatión (GSH) diminúe a curto prazo pero despois aumenta de novo.54
Heavy Metal Ions
Os ións metálicos pesados, como ferro, cobre, cadmio, mercurio, níquel, chumbo e arsénico, poden inducir a xeración de radicais reactivos e causar danos celulares mediante o esgotamento das actividades enzimáticas a través da peroxidación lipídica e a reacción con proteínas nucleares e DNA. 55
Un dos mecanismos máis importantes da xeración de radicais libres mediados por metais é a través dunha reacción de tipo Fenton. O ión superóxido e o peróxido de hidróxeno poden interactuar con metais de transición, como o ferro e o cobre, a través da reacción de Haber Weiss / Fenton catalizada polo metal para formar radicais OH.
Ademais dos mecanismos de tipo Fenton e Haber-Weiss, certos ións metálicos poden reaccionar directamente con moléculas celulares para xerar radicais libres, como radicais tiol, ou inducir vías de sinalización celular. Estes radicais tamén poden reaccionar con outras moléculas de tiol para xerar O22 .. O22. convértese en H2O2, o que provoca unha xeración adicional de radicais de osíxeno. Algúns metais, como o arsenito, inducen a formación de ROS indirectamente pola activación de sistemas produtores de radicais nas células
O arsénico é un elemento altamente tóxico que produce unha variedade de ROS, incluíndo superóxido (O2 2), osíxeno sinxelo (1O2), radical peroxilo (ROO), óxido nítrico (NO), peróxido de hidróxeno (H2O2) e radicais peroxilo dimetilarsínico [( CH3) 2AsOO] .57-59 Os compostos de arsénico (III) poden inhibir os encimas antioxidantes, especialmente os encimas dependentes da GSH, como glutatión-S-transferases (GST), glutatión peroxidasa (GSH-Px) e GSH redutase, mediante ligazón - dos seus grupos sulfhidrilo ((SH) .60,61
O chumbo aumenta a peroxidación de lípidos. 62 Disminúronse significativas na actividade do tecido SOD e cerebro GPx foron reportados logo da exposición ao chumbo. 63,64 O reemplazo do zinc, que serve como cofactor para moitas enzimas por plomo, leva á inactivación destes enzimas. A exposición ao chumbo pode causar inhibición do GST ao afectar os tióis do tecido.
Os ROS xerados por reaccións catalizadas por metais poden modificarse nas bases do ADN. Pódense producir tres substitucións de bases, G / C, G / T e C / T, como resultado do dano oxidativo por iones metálicos, como Fe21, Cu21 e Ni21. Reid et al65 mostraron que o G / C foi producido predominantemente por Fe21 mentres que a substitución C / T foi por Cu21 e Ni21.
ANTIOXIDANTES
O corpo humano está equipado cunha variedade de antioxidantes que serven para contrarrestar o efecto dos oxidantes. Para todos os efectos prácticos, estes poden dividirse en categorías 2: enzimática (Táboa 2) e nonenzimática (Táboa 3).
Antioxidantes enzimáticos
Os principais antioxidantes enzimáticos dos pulmóns son SOD (EC 1.15.1.11), catalase (EC 1.11.1.6) e GSH-Px (EC 1.11.1.9). Ademais destes enzimas importantes, tamén se atoparon outros antioxidantes, incluíndo heme oxigenase-1 (EC 1.14.99.3) e proteínas redox, como thioredoxins (TRXs, EC 1.8.4.10), peroxiredoxinas (PRXs, EC 1.11.1.15) e glutaredoxins. desempeñar papeis decisivos nas defensas antioxidantes pulmonares.
Dado que o superóxido é o ROS principal producido a partir dunha variedade de fontes, a súa dismutación por SOD é de importancia primordial para cada célula. Todas as formas 3 de SOD, isto é, CuZn-SOD, Mn-SOD e EC-SOD, son amplamente expresadas no pulmón humano. O Mn-SOD está localizado na matriz de mitocondrias. O EC-SOD está localizado principalmente na matriz extracelular, especialmente en áreas que conteñen altas cantidades de fibras de coláxeno de tipo I e en torno a vasos pulmonares e sistémicos. Tamén se detectou no epitelio bronquial, o epitelio alveolar e os macrófagos alveolares. 66,67 En xeral, considérase que xeralmente CuZn-SOD e Mn-SOD actúan como catalizadores masivos de radicais superóxidos. O nivel de EC-SOD relativamente alto no pulmón coa súa unión específica aos compoñentes da matriz extracelular pode representar un compoñente fundamental da protección da matriz pulmonar. 68
O H2O2 que se produce pola acción dos SOD ou a acción das oxidases, como a xantina oxidasa, é reducida ao auga por catalase e GSH-Px. A catalase existe como un tetraéser composto por monómeros idénticos 4, cada un deles contén un grupo hemo no sitio activo. A degradación do H2O2 realízase mediante a conversión entre conformacións 2 de catalase-ferricatalase (ferro coordinado ao auga) e composto I (ferro complexo cun átomo de osíxeno). Catalase tamén se une a NADPH como equivalente reductor para evitar a inactivación oxidativa da enzima (formación do composto II) por H2O2 xa que se reduce ao auga. 69
As enzimas do ciclo redox responsables da redución de H2O2 e os hidroperóxidos de lípidos (xerados como resultado da peroxidación de lípidos de membrana) inclúen a GSH-Pxs.70. Os GSH-Pxs son unha familia de enzimas tetrámeras que conteñen a única selenocisteína de aminoácidos dentro da Os sitios activos usan thiols de baixo peso molecular, como GSH, para reducir H2O2 e peróxidos de lípidos nos seus correspondentes alcohois. Catro GSH-Pxs foron descritos, codificados por diferentes xenes: GSH-Px-1 (GSH-Px celular) é omnipresente e reduce H2O2 e peróxidos de ácidos graxos, pero non hai peróxilos esterificados. 71 Os lípidos esterificados reducen o GSH unido a membranas -Px-4 (hidroperóxido fosfolípido GSH-Px), que pode usar varios tióoles de baixo peso molecular como equivalentes reductores. O GSH-Px-2 (GSH-Px gastrointestinal) localízase nas células epiteliales gastrointestinais onde serve para reducir os peróxidos dietéticos. 72 GSH-Px-3 (GSH-Px extracelular) é o único membro da familia GSH-Px que reside en o compartimento extracelular e crese que é un dos enzimas antioxidantes extracelulares máis importantes en mamíferos. Destes, o GSH-Px extracelular é máis amplamente investigado no pulmón humano. 73
Ademais, a eliminación de H2O2 está estrechamente asociada a varias enzimas que conteñen tiol, a saber, TRXs (TRX1 e TRX2), Thioredoxin reductases (EC 1.8.1.9) (TRRs), PRXs (que son peroxidases de thioredoxin) e glutaredoxinas. 74
Dous TRXs e TRRs caracterizáronse en células humanas, existentes tanto en citosol como en mitocondrias. No pulmón, TRX e TRR exprésanse en epitelio bronquial e alveolar e macrófagos. Se atoparon seis PRX diferentes nas células humanas, diferenciándose na súa compartimentación ultraestructural. Os estudos experimentais revelaron a importancia do PRX VI na protección do epitelio alveolar. O pulmón humano expresa todos os PRXs no epitelio bronquial, o epitelio alveolar e os macrófagos. 75 PRX V atopouse recentemente como unha peroxinitrite reductasa, 76, o que significa que pode funcionar como potencial composto protector no desenvolvemento da lesión pulmonar mediada por ROS. .77
Común a estes antioxidantes é o requisito de NADPH como equivalente reductor. NADPH mantén a catalase na forma activa e úsase como cofactor por TRX e GSH reductase (EC 1.6.4.2), que converte GSSG a GSH, un co-substrato para o GSH-Pxs. NADPH intracelular, á súa vez, xérase pola redución de NADP1 por glucosa-6-fosfato deshidroxenase, a primeira e limitante de limitación de velocidade da vía de fosfato de penosa, durante a conversión de glucosa-6-fosfato a 6-fosfogluconolactona. Ao xerar NADPH, a glucosa-6-fosfato deshidroxenase é un determinante crítico da capacidade tamponante de GSH citosólica (GSH / GSSG) e, polo tanto, pode considerarse unha enzima antioxidante reguladora esencial. 78,79
Os GST (EC 2.5.1.18), outra familia de encimas antioxidantes, inactivan metabolitos secundarios, como aldehídos insaturados, epóxidos e hidroperóxidos. Describíronse tres grandes familias de GST: GST citosólica, GST mitocondrial, 80,81 e GST microsómica asociada á membrana que ten un papel no metabolismo eicosanoide e GSH.82 Identifícanse sete clases de GST citosólica en mamíferos, denominada Alpha, Mu, Pi, Sigma, Theta, Omega e Zeta.83-86 Durante as condicións non estresadas, os GST de clase Mu e Pi interactúan con quinases Ask1 e JNK, respectivamente, e inhiben estas quinases. 87-89 Demostrouse que GSTP1 se disocia de JNK en resposta ao estrés oxidativo.89 O GSTP1 tamén interactúa fisicamente con PRX VI e leva á recuperación da actividade do encima PRX a través da glutatiónilación da proteína oxidada.90
Antioxidantes nonenzimáticos
Os antioxidantes nonenzimáticos inclúen compostos de baixo peso molecular, como vitaminas (vitaminas C e E), b-caroteno, ácido úrico e GSH, un tripéptido (Lg-glutamil-L-cisteinil-L-glicina) que comprende un tiol ( sulfhidrilo).
A vitamina C (ácido ascórbico)
A vitamina C (ácido ascórbico) soluble en auga proporciona a capacidade antioxidante en fase acuosa intracelular e extracelular principalmente por eliminar os radicales libres de osíxeno. Converte os radicales libres de vitamina E de volta á vitamina E. Os seus niveis plasmáticos demostraron que diminuír coa idade. 91,92
Vitamina E (a-tocoferol)
A vitamina E soluble en lípidos concentrábase no sitio interior hidrofóbico da membrana celular e é a principal defensa contra a lesión por membrana inducida por oxidación. A vitamina E dá electrón ao radical peroxilo, que se produce durante a peroxidación lipídica. a-Tocopherol é a forma máis activa de vitamina E e o principal antioxidante unido a membranas na célula. A vitamina E desencadea a apoptose das células cancerosas e inhibe as formacións de radicais libres. 93
Glutationa
O GSH é moi abundante en todos os compartimentos celulares e é o principal antioxidante soluble. A relación GSH / GSSG é un dos principais determinantes do estrés oxidativo. GSH mostra os seus efectos antioxidantes de varias formas. 94 Desintoxica o peróxido de hidróxeno e os peróxidos de lípidos a través da acción de GSH-Px. GSH doa o seu electrón a H2O2 para reducir a H2O e O2. GSSG volveuse a reducir a GSH por GSH reductase que usa NAD (P) H como o doador de electróns. Os GSH-Pxs tamén son importantes para a protección da membrana celular a partir da peroxidación lipídica. O glutatión reducido dá protones aos lípidos da membrana e os protexe contra ataques oxidantes. 95
O GSH é un cofactor para varias encimas desintoxicantes, como a GSH-Px ea transferase. Ten un papel na conversión de vitamina C e E de volta ás súas formas activas. GSH protexe as células contra a apoptose interactuando coas vías de sinalización proapoptótica e antiapoptótica.94 Tamén regula e activa varios factores de transcrición, como AP-1, NF-kB e Sp-1.
Carotenoides (b-caroteno)
Os carotenoides son pigmentos atopados nas plantas. Principalmente, atopouse que o b-caroteno reacciona cos radicales peroxil (ROO), hidroxilo (OH) e superóxido (O22). Os carotenoides 96 mostran os seus efectos antioxidantes na baixa presión parcial de osíxeno, pero poden ter efectos pro-oxidantes a un maior osíxeno concentracións.97 Tanto os carotenoides como os ácidos retinoicos (RA) son capaces de regular os factores de transcrición .98 b-Caroteno inhibe a activación de NF-kB inducida por oxidantes e a produción de interleucina (IL) -6 e factor de necrosis tumoral. Os carotenoides tamén afectan a apoptose das células. Os efectos antiproliferativos da RA mostráronse en varios estudos. Este efecto da RA está mediado principalmente por receptores de ácido retinoico e varía entre os tipos celulares. Nas células do carcinoma mamario, o receptor do ácido retinoico mostrou que provocou a inhibición do crecemento induciendo a prisión celular, a apoptose ou os dous. 99,100
O EFECTO DO ESTRÉS OXIDATIVO: MECANISMOS XENÉTICOS, FISIOLÓXICOS E BIOQUÍMICOS
O estrés oxidativo ocorre cando o equilibrio entre antioxidantes e ROS é interrompido debido ao esgotamento dos antioxidantes ou a acumulación de ROS. Cando se produce o estrés oxidativo, as células intentan contrarrestar os efectos oxidantes e restaurar o equilibrio redox mediante a activación ou silenciamiento de xenes que codifican encimas defensivos, factores de trangrafía e proteínas estruturais. A relación 101,102 entre o glutatión reducido e reducido (2GSH / GSSG) é un dos importantes determinantes do estrés oxidativo no corpo. A produción máis alta de ROS no corpo pode alterar a estrutura do ADN, provoca a modificación das proteínas e os lípidos, a activación de varios factores de transcrición inducida polo estrés e a produción de citoquinas proinflamatorias e antiinflamatorias.
Efectos do estrés oxidativo no ADN
O ROS pode levar ás modificacións do ADN de varias maneiras, o que implica degradación de bases, modificacións de ADN monocatenarias ou de dobre cadea, purinas, pirimidinas ou modificacións, mutacións, supresións ou translocaciones ligadas a azucre e enlaces cruzados con proteínas. A maioría destas modificacións de ADN (Fig. 1) son altamente relevantes para a carcinogénesis, envellecemento e enfermidades neurodegenerativas, cardiovasculares e autoinmunes. O fume de tabaco, os metais redox e os metais nonredox, como o ferro, o cadmio, o cromo eo arsénico, tamén están implicados na carcinogénesis e no envellecemento xerando radicais libres ou se unen a grupos tiol. A formación de 8-OH-G é o dano de ADN máis coñecido que se produce a través do estrés oxidativo e é un potencial biomarcador para a carcinogénesis.
As rexións promotoras de xenes conteñen secuencias de consenso para os factores de transcrición. Estes sitios de conexión de factores de transcrición conteñen secuencias ricas en GC que son susceptibles de ataques oxidantes. A formación de ADN 8-OH-G nos sitios de unión ao factor de transcrición pode modificar a unión dos factores de transcrición e cambiar así a expresión de xenes relacionados como se demostrou para as secuencias diana AP-1 e Sp-1. Tamén se demostrou que a 103-ciclo-8-desoxiadenosina (ciclo-dA) inhibe a transcrición dun xene reporteiro nun sistema celular se se atopa nunha caixa TATA.8,59 A proteína de unión a TATA inicia a transcrición cambiando a flexión do ADN . A unión da proteína de unión a TATA pode verse prexudicada pola presenza de ciclo-dA.
O estrés oxidativo provoca a inestabilidade das rexións de microsatélite (repeticións curtas en tándem). Os iones metálicos activos de Redox, os radicais hidroxilo aumentan a inestabilidade dos microsatélites. 105 Aínda que as pautas de ADN dunha soa hebra causadas por lesións oxidantes poden ser facilmente toleradas por células, as discontinuas de ADN bicatenario inducidas por radiacións ionizantes poden ser unha ameaza significativa para a supervivencia celular. 106
A metilación nas illas CpG no ADN é un importante mecanismo epigenético que pode producir silenciamento de xenes. Oxidación de 5-MeCyt para 5-hidroximetil uracilo (5-OHMeUra) pode ocorrer a través de reaccións de desaminação / oxidación de timina ou citosina 5-hidroximetil intermediates.107 Ademais da expresión do xene modulación, a metilação do ADN tamén parece afectar organization.108 cromatina Os patróns de metilación Aberrant de ADN inducidos por ataques oxidativos tamén afectan a actividade de reparación do ADN.
Efectos do estrés oxidativo nos lípidos
O ROS pode inducir a peroxidación de lípidos e interromper a disposición da bicapa lipídica da membrana que pode inactivar os receptores e encimas unidos á membrana e aumentar a permeabilidade dos tecidos.109 Os produtos da peroxidación de lípidos, como MDA e aldehídos insaturados, son capaces de inactivar moitas proteínas celulares formando cruz proteica. -ligas.110-112 4-hidroxi-2-nonenal provoca o esgotamento da GSH intracelular e induce a produción de peróxido, 113,114 activa o receptor do factor de crecemento epidérmico, 115 e induce a produción de fibronectina.116 Produtos de peroxidación de lípidos, como os isoprostanos e as sustancias reactivas do ácido tiobarbitúrico. , utilizáronse como biomarcadores indirectos de estrés oxidativo e mostráronse niveis aumentados no condensado respiratorio exhalado ou no fluído de lavado bronchoalveolar ou no pulmón de pacientes ou fumadores con enfermidade pulmonar obstructiva crónica.117-119
Efectos do estrés oxidativo nas proteínas
O ROS pode provocar a fragmentación da cadea peptídica, a alteración da carga eléctrica das proteínas, a reticulación das proteínas e a oxidación de aminoácidos específicos e, polo tanto, leva a unha maior susceptibilidade á proteólise por degradación por proteasas específicas. 120 Os residuos de cisteína e metionina nas proteínas son particularmente máis susceptibles á oxidación.121 A oxidación de grupos sulfhidrilo ou residuos de metionina das proteínas provoca cambios conformacionais, desdobramento das proteínas e degradación.8,121-123 Os encimas que teñen metais nos seus sitios activos ou próximos son especialmente máis sensibles á oxidación catalizada por metais. A modificación oxidativa dos encimas inhibe as súas actividades.124,125
Nalgúns casos, pode ocorrer a oxidación específica das proteínas. Por exemplo, a metionina pode ser oxidada de metionina sulfóxido 126 e fenilalanina á o-tirosina 127; Os grupos sulfhidrilo poden ser oxidados para formar enlaces disulfuro; 128 e grupos carbonilo poden ser introducidos nas redes laterais de proteínas. Os raios gamma, a oxidación catalizada por metal, o HOCl eo ozono poden causar a formación de grupos carbonilo. 129
Efectos do estrés oxidativo na transdución de sinais
A ROS pode inducir a expresión de varios xenes implicados na transducción de sinais.1,130 É importante unha alta relación entre GSH / GSSG para a protección da célula contra os danos oxidativos. A interrupción desta proporción provoca a activación de factores de transcrición sensibles ao redox, como NF-kB, AP-1, factor nuclear das células T activadas e factor inducible por hipoxia 1, que están implicados na resposta inflamatoria. A activación dos factores de transcrición a través de ROS conséguese mediante fervenzas de transducción de sinais que transmiten a información de fóra ao interior da célula. Os receptores da tirosina quinasa, a maioría dos receptores do factor de crecemento, como o receptor do factor de crecemento epidérmico, o receptor do factor de crecemento endotelial vascular e o receptor do factor de crecemento derivado de plaquetas, a proteína tirosina fosfatase e a serina / treonina quinases son obxectivos de ROS.131-133 As quinases reguladas polo sinal extracelular, JNK e p38, que son membros da familia das proteínas quinases activadas polo mitóxeno e implicadas en varios procesos nas células, incluíndo a proliferación, diferenciación e apoptose, tamén poden ser reguladas polos oxidantes.
En condicións de estrés oxidativo, os residuos de cisteína no sitio de unión ao ADN de c-Jun, algunhas subunidades AP-1 e a kB quinasa inhibitoria sofren S-glutathiolación reversible. A glutaredoxina e TRX xogaron un papel importante na regulación das vías de sinalización sensibles ao redox, como NF-kB e AP-1, a proteína quinasa p38 activada polo mitóxeno e JNK.134-137
NF-kB pódese activar en resposta a condicións de estrés oxidativo, como ROS, radicais libres e irradiación UV.138 A fosforilación de IkB libera NF-kB e permítelle entrar no núcleo para activar a transcrición dos xenes.139 Unha serie de quinases teñen informouse que fosforila IkB nos residuos de serina. Estas quinases son os obxectivos dos sinais oxidativos para a activación de NF-kB.140 Os axentes redutores melloran a unión ao ADN de NF-kB, mentres que os axentes oxidantes inhiben a unión ao ADN de NF-kB. TRX pode exercer dúas accións opostas na regulación de NF-kB: no citoplasma, bloquea a degradación de IkB e inhibe a activación de NF-kB pero mellora a unión ao ADN de NF-kB no núcleo.2 Activación de NF-kB mediante a degradación relacionada coa oxidación. de IkB resulta na activación de varios xenes relacionados coa defensa antioxidante. NF-kB regula a expresión de varios xenes que participan na resposta inmune, como IL-141b, IL-1, factor de necrose tumoral-a, IL-6 e varias moléculas de adhesión.8 NF-kB tamén regula a anxioxénese e proliferación e diferenciación das células.
O AP-1 tamén está regulado polo estado redox. Na presenza de H2O2, algúns iones metálicos poden inducir a activación de AP-1. Aumento da proporción de GSH / GSSG mellora a unión AP-1 mentres que o GSSG inhibe a unión ao DNA da unión do ADN de AP-1.144 do heterodímero Fos / Jun aumentada pola redución dunha única cisteína conservada no dominio de unión ao ADN de cada un dos As proteínas 145 mentres a unión ao ADN de AP-1 pode ser inhibida por GSSG en moitos tipos de células, o que suxire que a formación de enlaces disulfuro por residuos de cisteína inhibe a unión ao ADN-1. A transdución de sinais 146,147 mediante o estrés oxidativo resúmese na Figura 2.
CONCLUSIÓNS
O estrés oxidativo pode xurdir pola sobreproducción de ROS por reaccións metabólicas que usan osíxeno e cambian o equilibrio entre oxidante /antioxidante estados a favor dos oxidantes. Os ROS son producidos por actividades metabólicas celulares e por factores ambientais, como contaminantes atmosféricos ou fume de cigarro. Os ROS son moléculas altamente reactivas debido a electróns non aparelados na súa estrutura e reaccionan con varias macromoléculas biolóxicas en células, como carbohidratos, ácidos nucleicos, lípidos e proteínas e alteran as súas funcións. O ROS tamén afecta a expresión de varios xenes mediante a regulación dos factores de transcrición sensibles a redox e a remodelación da cromatina por alteración na acetilación / desacetilación de histonas. A regulación do estado redox é fundamental para a viabilidade celular, a activación, a proliferación e a función do órgano.
Referencias
1. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Radicais libres, metais e antioxidantes no cancro inducido polo estrés oxidativo. Chem Biol Interact. 2006; 160: 1 40.
2. Halliwell B, Gutteridge JMC. Radicais libres en bioloxía e medicina. 3rd ed. Nova York: Oxford University Press; 1999.
3. Marnett LJ. Peroxidación de lípidos e danos no ADN por malondialdehído. Mutat Res. 1999; 424: 83-95.
4. Siems WG, Grune T, Esterbauer H. Formación de 4-hidroxinonenal durante a isquemia e a reperfusión do intestino delgado de rata. � Ciencia da vida. 1995;57:785-789.
5. Stadtman ER. Papel das especies oxidantes no envellecemento. Curr Med Chem. 2004; 11: 1105-1112.
6. Wang MY, Dhingra K, Hittelman WN, Liehr JG, deAndrade M, Li DH. O ADN de malondialdehído suposto inducido pola peroxidación lipídica-ADN nos tecidos mamarios humanos. Biomarcadores do epidemiol do cancro Prev. 1996; 5: 705-710.
7. Jenner P. Estrés oxidativo na enfermidade de Parkinson. Ann Neurol. 2003; 53: S26 S36.
8. Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. Unha avaliación do dano oxidativo a proteínas, lípidos e ADN no cerebro de pacientes con enfermidade de Alzheimer. J Neurochem. 1997; 68: 2061-2069.
9. Sayre LM, Smith MA, Perry G. Química e bioquímica do estrés oxidativo en enfermidades neurodexenerativas. Curr Med Chem. 2001; 8: 721-738.
10. Toshniwal PK, Zarling EJ. Evidencias dun aumento da peroxidación lipídica na esclerose múltiple. Neurochem Res. 1992; 17: 205-207.
11. Dhalla NS, Temsah RM, Netticadan T. Papel do estrés oxidativo en enfermidades cardiovasculares. J Hipertensos. 2000; 18: 655-673.
12. Kasparova S, Brezova V, Valko M, Horecky J, Mlynarik V, et al. Estudo do estrés oxidativo nun modelo de rata de hipoperfusión cerebral crónica. Neurochem Int. 2005; 46: 601-611.
13. Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. A produción de anións superóxido aumenta nun modelo de hipertensión xenética: o papel do endotelio. Hipertensión. 1999; 33: 1353-1358.
14. Kukreja RC, Hess ML. O sistema de radicais libres de osíxeno: desde ecuacións a través das interaccións proteínicas da membrana ata lesións e protección cardiovascular. Cardiovasc Res. 1992; 26: 641 655.
15. Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, et al. O tabaquismo induce un aumento do dano oxidativo no ADN, a 8-hidroxidoxiguanosina, nun sitio central do pulmón humano. Carcinoxénese. 1997; 18: 1763-1766.
16. Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Acontecementos oxidativos e nitrosativos na asma. Radical libre Biol Med. 2003; 35: 213-225.
17. Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA, et al. Correlación da deficiencia sistémica de superóxido dismutase coa obstrución do fluxo de aire no asma. Am J Respir Crit Care Med. 2005; 172: 306-313.
18. Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A, et al. Inactivación da superóxido dismutase en fisiopatoloxía da remodelación e reactividade das vías respiratorias asmáticas. Son J Pathol. 2005; 166: 663.
19. Dut R, Dizdar EA, Birben E, Sackesen C, Soyer OU, Besler T, Kalayci O. O estrés oxidativo e os seus determinantes nas vías aéreas de nenos con asma. Alerxia. 2008; 63: 1605-1609.
20. Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, et al. Estrés oxidativo e determinantes xenéticos e epidemiolóxicos da lesión oxidante no asma infantil. Clinica de alerxia J Immunol. 2006; 118: 1097-1104.
21. Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Programa de investigación sobre asma grave. A homeostase do glutatión das vías respiratorias está alterada en nenos con asma grave: evidencia de estrés oxidante. Clinica de alerxia J Immunol. 2009; 123: 146-152.
22. Miller DM, Buettner GR, Aust SD. Os metais de transición como catalizadores de reaccións de "autoxidación". Radical libre Biol Med. 1990; 8: 95-108.
23. Dupuy C, Virion A, Ohayon R, Kaniewski J, Dme D, Pommier J. Mecanismo de formación de peróxido de hidróxeno catalizado pola NADPH oxidasa na membrana plasmática da tiroide. J Biol Chem. 1991; 266: 3739-3743.
24. DN Granger. Papel da xantina oxidase e dos granulocitos na lesión por isquemiaperfusión. Son J Physiol. 1988; 255: H1269 H1275.
25. Fenton HJH. Oxidación do ácido tartárico en presenza de ferro. J Chem Soc. 1984; 65: 899-910.
26. Haber F, Weiss JJ. A descomposición catalítica do peróxido de hidróxeno por sales de ferro. Proc R Soc Lond Ser A. 1934; 147: 332 351.
27. Liochev SI, Fridovich I. The Haber Weiss ciclou 70 anos despois: unha visión alternativa. Redox Rep.2002; 7: 55-57.
28. Klebanoff SJ. Mieloperoxidase: amigo e inimigo. J Leukoc Biol. 2005; 77: 598-625.
29. Whiteman M, Jenner A, Halliwell B. Modificacións de bases inducidas por ácido hipocloroso no ADN do timo do becerro illado. Chem Res Toxicol. 1997; 10: 1240-1246.
30. Kulcharyk PA, Heinecke JW. O ácido hipocloroso producido polo sistema mieloperoxidase dos fagocitos humanos induce enlaces cruzados covalentes entre o ADN e a proteína. Bioquímica. 2001; 40: 3648-3656.
31. Brennan ML, Wu W, Fu X, Shen Z, Song W, et al. Unha historia de dúas controversias: definir o papel das peroxidasas na formación de nitrotirosina in vivo usando ratos eosinófilos peroxidases e mieloperoxidas e a natureza das especies reactivas de nitróxeno xeradas por peroxidasa. J Biol Chem. 2002; 277: 17415-17427.
32. Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, et al. A desgranulación dos eosinófilos e a oxidación mediada pola peroxidasa das proteínas das vías respiratorias non ocorren nun modelo de inflamación pulmonar que supón un ovalbúmina de rato. J Immunol. 2001; 167: 1672-1682.
33. van Dalen CJ, Winterbourn CC, Senthilmohan R, Kettle AJ. O nitrito como substrato e inhibidor da mieloperoxidase. Implicacións para a nitración e a produción de ácido hipocloroso nos sitios de inflamación. J Biol Chem. 2000; 275: 11638 11644.
34. Wood LG, Fitzgerald DA, Gibson PG, Cooper DM, Garg ML. A peroxidación lipídica determinada polos isoprostanos plasmáticos está relacionada coa gravidade da enfermidade no asma leve. Lípidos. 2000; 35: 967 974.
35. Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. Aumento do 8-isoprostano, un marcador de estrés oxidativo, no condensado exhalado de pacientes con asma. Am J Respir Crit Care Med. 1999; 160: 216-220.
36. Igrexa DF, Pryor WA. Química de radicais libres do fume do cigarro e as súas implicacións toxicolóxicas. Perspectiva da saúde ambiental. 1985; 64: 111-126.
37. Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ, et al. Inflamación inducida polo ozono avaliada en esputo e fluído de lavado bronquial de asmáticos: unha nova ferramenta non invasiva en estudos epidemiolóxicos sobre contaminación atmosférica e asma. Radical libre Biol Med. 1999; 27: 1448-1454.
38. Nightingale JA, Rogers DF, Barnes PJ. Efecto do ozono inhalado sobre o óxido nítrico exhalado, a función pulmonar e o esputo inducido en suxeitos normais e asmáticos. Tórax. 1999; 54: 1061-1069.
39. Cho AK, Sioutas C, Miguel AH, Kumagai Y, Schmitz DA, et al. Actividade redox de partículas en suspensión no aire en diferentes sitios da conca dos Ánxeles. Res Res. 2005; 99: 40-47.
40. Comhair SA, Thomassen MJ, Erzurum SC. Indución diferencial da glutationa peroxidasa extracelular e do óxido nítrico sintase 2 nas vías respiratorias de individuos sans expostos ao 100% O (2) ou fume de cigarro. Am J Respir Cell Mol Biol. 2000; 23: 350-354.
41. Matthay MA, Geiser T, Matalon S, Ischiropoulos H. Lesión pulmonar mediada por oxidante na síndrome de angustia respiratoria aguda. Crit Care Med. 1999; 27: 2028-2030.
42. Biaglow JE, Mitchell JB, Held K. A importancia do peróxido e superóxido na resposta de raios X. Int J Radiat Oncol Biol Phys. 1992; 22: 665-669.
43. Chiu SM, Xue LY, Friedman LR, Oleinick NL. Sensibilización mediada por ións de cobre dos sitios de unión da matriz nuclear á radiación ionizante. Bioquímica. 1993; 32: 6214.
44. Narayanan PK, Goodwin EH, Lehnert BE. As partículas alfa inician a produción biolóxica de anións superóxidos e peróxido de hidróxeno nas células humanas. Res Cancro. 1997; 57: 3963-3971.
45. Tuttle SW, Varnes ME, Mitchell JB, Biaglow JE. Sensibilidade aos oxidantes químicos e á radiación nas liñas celulares CHO deficientes na actividade do ciclo da pentosa oxidativa. Int J Radiat Oncol Biol Phys. 1992; 22: 671-675.
46. Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, e outros. Manganeso
expresión xénica mediada por disolución de superóxido dismutase en radiación inducida
respostas adaptativas. Mol Cell Biol. 2003; 23: 2362 - 2378.
47. Azzam EI, de Toledo SM, Spitz DR, Little JB. Metabolismo oxidativo
modula a transdución de sinais ea formación de micronúcleos no espectador
As células a partir de partículas irradian fibroblastos humanos normais. Cancer Res.
2002; 62: 5436-5442.
48. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB.
Xeración reactiva de mitocondrias inducida por radiacións ionizantes
osíxeno / nitróxeno. Res Cancro. 2001; 61: 3894-3901.
49. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK rutas en
respostas de radiación. Oncoxene. 2003; 22: 5885-5896.
50. Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, e outros. Tioredoxina
a translocación nuclear e a interacción co factor redox-1 activa o factor de transcrición AP-1 en resposta á radiación ionizante. Res Cancro. 2000; 60: 6688 - 6695.
51. Cadete J, Douki T, Gasparutto D, Ravanat JL. Dano oxidativo no ADN: formación, medida e características bioquímicas. Mutat Res. 2003; 531: 5.
52. Yokoya A, Cunniffe SM, O Neill P. Efecto da hidratación na indución de roturas de cadeas e lesións de base en películas de ADN plásmido por gammaradiación. J Am Chem Soc. 2002; 124: 8859 8866.
53. Janssen YM, Van Houten B, Borm PJ, Mossman BT. Respostas das células e dos tecidos ao dano oxidativo. Laboratorio Invest. 1993; 69: 261-274.
54. Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T, et al. Indución dependente do factor nuclear kappa B da gamma glutamilcisteína sintetase por radiación ionizante nas células do glioblastoma humano T98G. Radical libre Biol Med. 1998; 24: 1256-1268.
55. Stohs SJ, Bagchi D. Mecanismos oxidativos na toxicidade dos ións metálicos. Radical libre Biol Med. 1995; 18: 321-336.
56. Leonard SS, Harris GK, Shi X. Estrés oxidativo inducido polo metal e transducción de sinais. Radical libre Biol Med. 2004; 37: 1921-1942.
57. Shi H, Shi X, Liu KJ. Mecanismo oxidativo da toxicidade e carcinoxénese do arsénico. Mol Cell Biochem. 2004; 255: 67-78.
58. Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T. Un mecanismo potencial para o deterioro da formación de óxido nítrico causado pola exposición oral prolongada ao arsenato nos coellos. Free Radic Biol Med.2003; 35: 102-113.
59. Rin K, Kawaguchi K, Yamanaka K, Tezuka M, Oku N, Okada S. As roturas de DNAstrand inducidas polo ácido dimetilarsínico, un metabolito do arsénico inorgánico, están fortemente reforzadas polos radicais aniónicos superóxidos. Biol Pharm Bull. 1995; 18: 45-58.
60. Deputado de Waalkes, Liu J, Ward JM, Diwan LA. Mecanismos subxacentes á carcinoxénese do arsénico: hipersensibilidade dos ratos expostos ao arsénico inorgánico durante a xestación. Toxicoloxía. 2004; 198: 31-38.
61. Schiller CM, Fowler BA, Woods JS. Efectos do arsénico na activación da piruvato deshidroxenase. Perspectiva da saúde ambiental. 1977; 19: 205-207.
62. Monterio HP, Bechara EJH, Abdalla DSP. Participación de radicais libres en porfirias neurolóxicas e envelenamento por chumbo. Mol Cell Biochem. 1991; 103: 73–83.
63. Tripathi RM, Raghunath R, Mahapatra S. Chumbo no sangue e o seu efecto sobre os niveis de Cd, Cu, Zn, Fe e hemoglobina dos nenos. Sci Total Environ. 2001; 277: 161-168.
64. Nehru B, Dua R. O efecto do selenio na dieta sobre a neurotoxicidade do chumbo. J Environ Pathol Toxicol Oncol. 1997; 16: 47-50.
65. Reid TM, Feig DI, Loeb LA. Mutaxénese por radicais de osíxeno inducidos por metais. Perspectiva de saúde ambiental. 1994; 102 (supl. 3): 57 61.
66. Kinnula VL, Crapo JD. Superóxido dismutase nas enfermidades pulmonares e pulmonares humanas. Am J Respir Crit Care Med. 2003; 167: 1600-1619.
67. Kinnula VL. Produción e degradación de metabolitos de osíxeno durante estados inflamatorios no pulmón humano. As drogas Curr están dirixidas á alerxia á inflamación. 2005; 4: 465-470.
68. Zelko IN, Mariani TJ, Folz RJ. Familia multíxeno de superóxido dismutase: unha comparación das estruturas, evolución e expresión xénica do CuZn-SOD (SOD1), Mn-SOD (SOD2) e EC-SOD (SOD3). Radical libre Biol Med. 2002; 33: 337-349.
69. Kirkman HN, Rolfo M, Ferraris AM, Gaetani GF. Mecanismos de protección da catalase por NADPH. Cinética e estequiometría. J Biol Chem. 1999; 274: 13908-13914.
70. Floh L. Glutatión peroxidasa. Ciencias básicas da vida. 1988; 49: 663-668.
71. Arthur JR. As glutatione peroxidasas. Cell Mol Life Sci. 2000; 57: 1825-1835.
72. Chu FF, Doroshow JH, Esworthy RS. Expresión, caracterización e distribución tisular dunha nova glutatión peroxidasa dependente do selenio, GSHPx-GI. J Biol Chem. 1993; 268: 2571-2576.
73. Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Indución extracelular de glutatión peroxidasa en pulmóns asmáticos: evidencia da regulación redox da expresión nas células epiteliais das vías respiratorias humanas. FASEB J. 2001; 15: 70-78.
74. Gromer S, Urig S, Becker K. O sistema de tioredoxina da ciencia á clínica. Med Res Rev. 2004; 24: 40–89.
75. Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, P kk P, et al. Expresión específica celular de peroxiredoxinas na sarcoidose pulmonar e pulmonar humana. Tórax. 2002; 57: 157-164.
76. Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T, et al. A peroxiredoxina 5 humana é unha peroxinitrita redutase. FEBS Lett. 2004; 571: 161-165.
77. Holmgren A. Función antioxidante dos sistemas de tioredoxina e glutaredoxina. Sinal redox antioxidante. 2000; 2: 811-820.
78. Dickinson DA, Forman HJ. Glutatión en defensa e sinalización: leccións dun pequeno tiol. Ann NY Acad Sci. 2002; 973: 488-504.
79. Sies H. Glutatión e o seu papel nas funcións celulares. Radical libre Biol Med. 1999; 27: 916-921.
80. Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. Vías evolutivas paralelas para glutatión transferases: estrutura e mecanismo do encima kappa da clase mitocondrial rGSTK1-1. Bioquímica. 2004; 43: 52 61.
81. Robinson A, Huttley GA, Booth HS, Board PG. Os estudos de modelado e bioinformática da clase kappa humana glutatión transferase predicen unha nova terceira familia de transferases con homoloxía ás isomerases 2-hidroxicrómenos-2-carboxilatos procariotas. Biochem J. 2004; 379: 541-552.
82. Jakobsson PJ, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Características estruturais comúns de MAPEGda superfamilia estendida de proteínas asociadas á membrana con funcións altamente diverxentes no metabolismo eicosanoide e do glutatión. Proteínas Sci. 1999; 8: 689.
83. Hayes JD, Pulford DJ. A familia de superxenos glutatión S-transferasa: regulación da GST e contribución dos isoenzimas á quimioprotección do cancro e á resistencia aos medicamentos. Crit Rev Biochem Mol Biol. 1995; 30: 445-600.
84. Armstrong RN. Estrutura, mecanismo catalítico e evolución das glutatión transferases. Chem Res Toxicol. 1997; 10: 2-18.
85. Hayes JD, McLellan LI. Os encimas dependentes do glutatión e do glutatión representan unha defensa regulada contra o estrés oxidativo. Res. Radicais libres. 1999; 31: 273-300.
86. Sheehan D, Meade G, Foley VM, Dowd CA. Estrutura, función e evolución das glutatión transferases: implicacións para a clasificación de membros non mamíferos dunha antiga superfamilia enzimática. Biochem J. 2001; 360: 1-16.
87. Cho SG, Lee YH, Park HS, Ryoo K, Kang KW, et al. A glutatión S-transferase Mu modula os sinais activados polo estrés suprimindo a quinasa reguladora do sinal da apoptose 1. J Biol Chem. 2001; 276: 12749.
88. Dorion S, Lambert H, Landry J. A activación da vía de sinalización p38 por choque térmico implica a disociación da glutatión S-transferasa Mu de Ask1. J Biol Chem. 2002; 277: 30792-30797.
89. Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, et al. Regulación da sinalización JNK por GSTp. EMBO J. 1999; 18: 1321-1334.
90. Manevich Y, Feinstein SI, Fisher AB. A activación do encima antioxidante 1-CYS peroxiredoxina require glutatiónilación mediada por heterodimerización con pGST. Proc Natl Acad Sci EU A. 2004; 101: 3780-3785.
91. Bunker VW. Radicais libres, antioxidantes e envellecemento. Med Lab Sci. 1992; 49: 299-312.
92. Mezzetti A, Lapenna D, Romano F, Costantini F, Pierdomenico SD, et al. O estrés oxidativo sistémico e a súa relación coa idade e a enfermidade. J Am Geriatr Soc. 1996; 44: 823-827.
93. White E, Shannon JS, Patterson RE. Relación entre vitamina e
uso de suplementos de calcio e cancro de colon. Biomarcadores do epidemiol do cancro Prev. 1997; 6: 769 774.
94. Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Novos mecanismos de compostos antioxidantes naturais en sistemas biolóxicos: implicación de glutatión e encimas relacionados co glutatión. J Nutr Bioquímica. 2005; 16: 577-586.
95. Curello S, Ceconi C, Bigoli C, Ferrari R, Albertini A, Guarnieri C. Cambios no estado de glutatión cardíaco despois da isquemia e a reperfusión. Experientia. 1985; 41: 42-43.
96. El-Agamey A, Lowe GM, McGarvey DJ, Mortensen A, Phillip DM, Truscott TG. Química radical radical carotenoide e propiedades antioxidantes / pro-oxidantes. Arch Biochem Biophys. 2004; 430: 37-48.
97. Rice-Evans CA, Sampson J, Bramley PM, Holloway DE. Por que esperamos que os carotenoides sexan antioxidantes in vivo? Res. Radicais libres. 1997; 26: 381-398.
98. Niles RM. Vías de sinalización na quimioprevención retinoide e tratamento do cancro. Mutat Res. 2004; 555: 81-96.
99. Donato LJ, Noy N. Supresión do crecemento do carcinoma mamario por parte do ácido retinoico: os xenes proapoptóticos son obxectivos da sinalización do receptor de ácido retinoico e da proteína II de unión ao ácido retinoico celular. Res Cancro. 2005; 65: 8193-8199.
100. Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, et al. O Bcl-2 é un regulador clave para a morte de células apoptóticas inducidas por ácido retinoico no neuroblastoma. Oncoxene. 2006; 25: 5046 - 5055.
101. Dalton TP, Shertzer HG, Puga A. Regulación da expresión xénica mediante osíxeno reactivo. Ann Rev Pharmacol Toxicol. 1999; 39: 67-101.
102. Scandalios JG. Respostas xenómicas ao estrés oxidativo. En: Meyers RA, ed. Enciclopedia de Bioloxía Celular Molecular e Medicina Molecular. Vol 5. 2a ed. Weinheim, Alemaña: Wiley-VCH; 2004: 489-512.
103. Ghosh R, Mitchell DL. Efecto do dano oxidativo no ADN nos elementos promotores na unión ao factor de transcrición. Ácidos nucleicos Res. 1999; 27: 3213-3218.
104. Marietta C, Gulam H, Brooks PJ. Unha única lesión de 8, 50-ciclo-20-desoxiadenosina nunha caixa TATA impide a unión da proteína de unión TATA e reduce fortemente a transcrición in vivo. Reparación do ADN (Amst). 2002; 1: 967-975.
105. Jackson AL, Chen R, Loeb LA. Inducción da inestabilidade microsatélica
por danos no ADN oxidativo. Proc Natl Acad Sci US A. 1998; 95: 12468-12473.
106. Caldecott KW. Interaccións proteína-proteína durante a reparación de rotura dunha cadea de ADN de mamíferos. Biochem Soc Trans. 2003; 31: 247-251.
107. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Dano ao ADN oxidativo: mecanismos, mutación e enfermidade. FASEB J. 2003; 17: 1195–1214.
108. Jones PL, Wolffe AP. Relacións entre a organización da cromatina e a metilación do ADN na determinación da expresión xénica. Semin Cancer Biol. 1999; 9: 339-347.
109. Girotti AW. Mecanismos de peroxidación lipídica. J Free Radic Biol Med. 1985; 1: 87-95.
110. Siu GM, Draper HH. Metabolismo do malonaldehido in vivo e in vitro. Lípidos. 1982; 17: 349 355.
111. Esterbauer H, Koller E, Slee RG, Koster JF. Posible implicación do produto de peroxidación de lípidos 4-hidroxinonenal na formación de cromolípidos fluorescentes. Biochem J. 1986; 239: 405-409.
112. Hagihara M, Nishigaki I, Maseki M, Yagi K. Cambios dependentes da idade nos niveis de peróxido de lípidos nas fraccións lipoproteínicas do soro humano. J Gerontol. 1984; 39: 269-272.
113. Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, et al. 4- O hidroxinonenal, un produto aldehídico da peroxidación dos lípidos da membrana, prexudica o transporte de glutamato e a función mitocondrial nos sinaptosomas. Neurociencia. 1997; 806: 85-96.
114. Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activación das vías de sinalización do estrés polo produto final da peroxidación lipídica. O 4-hidroxi-2-nonenal é un indutor potencial da produción de peróxido intracelular. J Biol Chem. 1999; 274: 2234-2242.
115. Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jurgens G, Salvayre R, Negre-Salvayre A. Activación do receptor EGF por LDL oxidado. FASEB J. 1998; 12: 665-671.
116. Tsukagoshi H, Kawata T, Shimizu Y, Ishizuka T, Dobashi K, Mori M. 4-Hydroxy-2-nonenal mellora a produción de fibronectina polos fibroblastos de pulmón humano IMR-90 en parte a través da activación do sinal extracelular ligado ao receptor do factor de crecemento epidérmico- vía cinasa regulada p44 / 42. Toxicol Appl Pharmacol. 2002; 184: 127-135.
117. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. O 8-isoprostano expirado como biomarcador in vivo do estrés oxidativo pulmonar en pacientes con EPOC e fumadores sans. Am J Respir Crit Care Med. 2000; 162: 1175-1177.
118. Morrison D, Rahman I, Lannan S, MacNee W. Permeabilidade epitelial, inflamación e estrés oxidante nos espazos aéreos dos fumadores. Am J Respir Crit Care Med. 1999; 159: 473-479.
119. Nowak D, Kasielski M, Antczak A, Pietras T, Bialasiewicz P. Aumento do contido de substancias reactivas ao ácido tiobarbitúrico e peróxido de hidróxeno no condensado respiratorio caducado de pacientes con enfermidade pulmonar obstructiva crónica estable: sen efecto significativo do tabaquismo. Respir Med. 1999; 93: 389-396.
120. Kelly FJ, Mudway IS. Oxidación de proteínas na interface aire-pulmón. Aminoácidos. 2003; 25: 375-396.
121. Dean RT, Roberts CR, Jessup W. Fragmentación de polipéptidos extracelulares e intracelulares por radicais libres. Prog Clin Biol Res. 1985; 180: 341-350.
122. Keck RG. O uso de hidroperóxido de t-butilo como sonda para a oxidación da metionina nas proteínas. Bioquímica anal. 1996; 236: 56-62.
123. Davies KJ. Dano e degradación das proteínas por radicais de osíxeno. I. Aspectos xerais. J Biol Chem. 1987; 262: 9895-9901.
124. Stadtman ER. Oxidación catalizada por ións metálicos das proteínas: mecanismo bioquímico e consecuencias biolóxicas. Gratis Radic Biol Med.
1990; 9: 315-325.
125. Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inactivación de encimas metabólicos clave por reaccións de oxidación de función mixta: posible implicación no cambio de proteínas e o envellecemento. Proc Natl Acad Sci US A. 1983; 80: 1521-1525.
126. Stadtman ER, Moskovitz J, Levine RL. Oxidación dos residuos de metionina das proteínas: consecuencias biolóxicas. Sinal redox antioxidante. 2003; 5: 577-582.
127. Stadtman ER, Levine RL. Oxidación mediada por radicais libres de aminoácidos libres e residuos de aminoácidos nas proteínas. Aminoácidos. 2003; 25: 207-218.
128. Stadtman ER. Oxidación de proteínas no envellecemento e enfermidades relacionadas coa idade. Ann NY Acad Sci. 2001; 928: 22-38.
129. Shacter E. Cuantificación e importancia da oxidación de proteínas en mostras biolóxicas. Drug Metab Rev. 2000; 32: 307-326.
130. Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Estrés oxidativo e sinalización celular. Curr Med Chem. 2004; 11: 1163-1182.
131. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Factor de crecemento endotelial vascular (VEGF) e os seus receptores. FASEB J. 1999; 13: 9 22.
132. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, et al. Regulación da xeración de especies de osíxeno reactivo en fibroblastos por Rac1. Biochem J. 1996; 318: 379-382.
133. Sun T, Oberley LW. Regulación redox de activadores transcricionais. Radical libre Biol Med. 1996; 21: 335-348.
134. Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Regulación redox da unión ao ADN c-Jun por S-glutathiolación reversible. FASEB J. 1999; 13: 1481-1490.
135. Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen Heininger
YM. Detección in situ de proteínas S-glutationiladas tras a derivatización da cisteína catalizada por glutaredoxina-1. Biochim Biophys Acta. 2006; 1760: 380-387.
136. Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulación de glutaredoxina-1
expresión nun modelo de rato de enfermidade alérxica das vías respiratorias. Am J Respir Cell Mol Biol. 2007; 36: 147-151.
137. Filomeni G, Rotilio G, Ciriolo MR. A sinalización celular e o sistema redox de glutatión. Biochem Pharmacol. 2002; 64: 1057-1064.
138. Pande V, Ramos MJ. Recoñecemento molecular da 15-desoxidelta (12,14) prostaglandina J (2) polo factor nuclear-kappa B e outras proteínas celulares. Bioorg Med Chem Lett. 2005; 15: 4057-4063.
139. Perkins ND. Integración de vías de sinalización celular coa función NF-kappaB e IKK. Nat Rev Mol Cell Biol. 2007; 8: 49 ́62.
140. Gilmore TD. Introdución a NF-kappaB: xogadores, vías, perspectivas. Oncoxene. 2006; 25: 6680-6684.
141. Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Papeis distintos da tioredoxina no citoplasma e no núcleo. Un mecanismo en dous pasos de regulación redox do factor de transcrición NF-kappaB. J Biol Chem. 1999; 274: 27891-27897.
142. Ward PA. Papel do complemento, quimiocinas e citocinas reguladoras na lesión pulmonar aguda. Ann NY Acad Sci. 1996; 796: 104-112.
143. Akira S, Kishimoto A. NF-IL6 e NF-kB na regulación xénica das citocinas. Adv Immunol. 1997; 65: 1-46.
144. Meyer M, Schreck R, Baeuerle PA. O H2O2 e os antioxidantes teñen efectos opostos na activación de NF-kappa B e AP-1 en células intactas: AP-1 como factor secundario que responde aos antioxidantes. EMBO J. 1993; 12: 2005-2015.
145. Abate C, Patel L, Rausher FJ, Curran T. Regulación redox da actividade de unión do ADN fos e jun in vitro. Ciencia. 1990; 249: 1157-1161.
146. Galter D, Mihm S, Droge W. Efectos distintos do disulfuro de glutatión sobre os factores de transcrición nuclear kB e a proteína activadora-1. Eur J Biochem. 1994; 221: 639-648.
147. Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. A actividade transcricional AP-1 está regulada por unha asociación directa entre tioredoxina e Ref-1. Proc Natl Acad Sci US A. 1997; 94: 3633-3638.
A ferramenta Find A Practitioner de IFM é a rede de referencia máis grande en Medicina Funcional, creada para axudar aos pacientes a localizar a profesionais da Medicina Funcional en calquera parte do mundo. Os profesionais certificados IFM aparecen en primeiro lugar nos resultados da busca, dada a súa ampla educación en Medicina Funcional